
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed asymmetric intermolecular Mizoroki–Heck reaction for construction of a chiral quaternary carbon center†
Qing-Song
Zhang
a,
Shi-Li
Wan
a,
Di
Chen
a,
Chang-Hua
Ding
*a and
Xue-Long
Hou
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: dingch@sioc.ac.cn; xlhou@sioc.ac.cn; Tel: +86 21 5492 5144
bShanghai–Hong Kong Joint Laboratory in Chemical Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 12th June 2015
Abstract
Palladium-catalyzed asymmetric intermolecular Mizoroki–Heck reaction for the construction of a chiral quaternary carbon center is developed, affording 2,2-disubstituted 2,5-dihydrofurans in high yield with excellent enantioselectivity. The products are easily converted into the corresponding butenolides with retention of enantioselectivity.
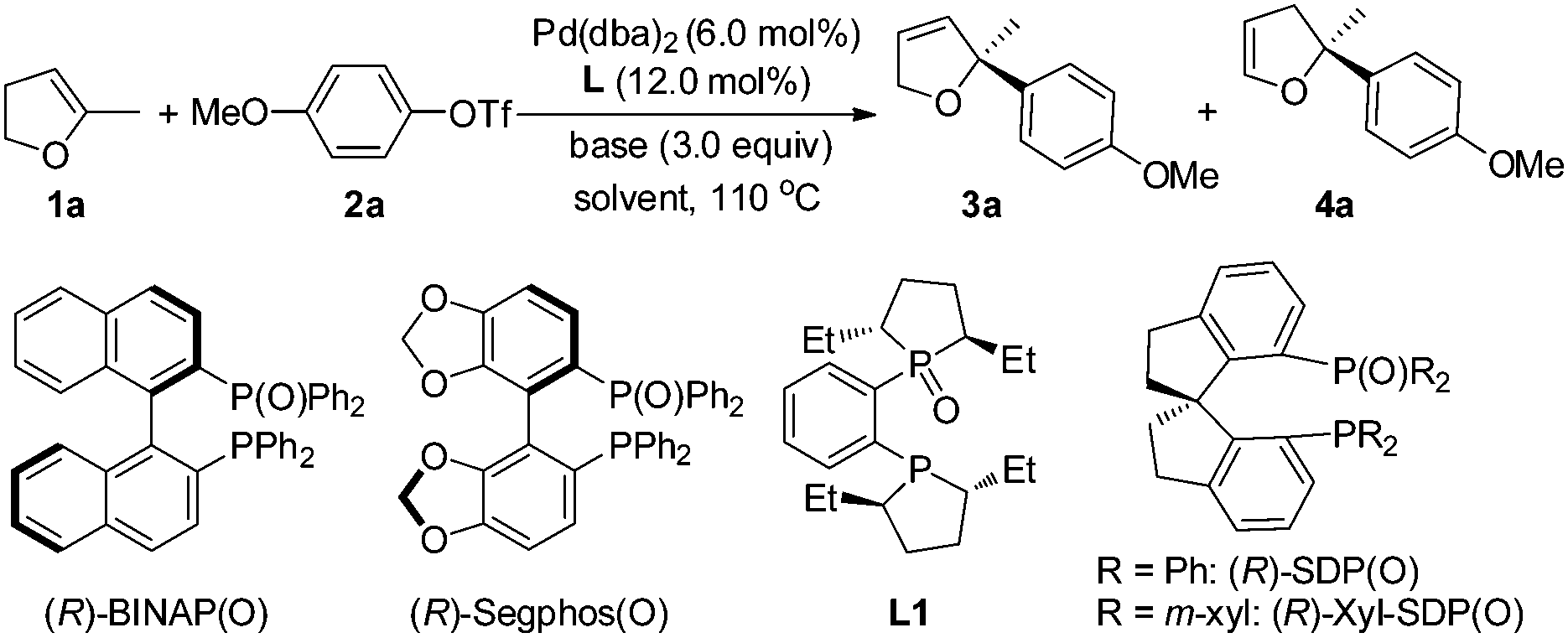
The Pd-catalyzed intermolecular Mizoroki–Heck reaction between olefins and organic halides has become one of the most powerful tools for C–C bond formation and found a wide range of applications in the synthesis of natural products, pharmaceuticals, and functional materials since its discovery more than 40 years ago.1 Twenty years later, the first intermolecular asymmetric variant was reported by Hayashi and co-workers.2 This pioneering work has inspired many groups to devote considerable efforts to this area.3 However, only a limited number of substrates were suitable for this Pd-catalyzed asymmetric intermolecular reaction for a long time, which hampered the applications of the reaction in organic synthesis severely. Only recently, some significant breakthroughs have been made by Jung, Zhou and Sigman.3e,4–6 Aryl halides and benzylic electrophiles have been used successfully in Pd-catalyzed asymmetric intermolecular Mizoroki–Heck reaction, affording the corresponding products in high enantioselectivities.5 Even acyclic alkenes as well as trisubstituted alkenes have also been used in oxidative Heck or Heck-Matsada reaction to form a chiral quaternary carbon center with excellent efficiency regarding the yield and enantioselectivity.6f These newly developed methodologies have also found their applications in organic synthesis.5b,c,6c In spite of the great progress that has been achieved, successful examples are still limited with rather narrow substrate scope, especially the reports concerning the use of trisubstituted alkenes and the methodology to form a chiral quaternary carbon center, one of the key issues in asymmetric synthesis, are very few.6f,7 Only one example of Pd-catalyzed asymmetric Heck reaction of 5-methyl-2,3-dihydrofuran with PhOTf was given by Tschoerner and Pregosin, affording the corresponding Heck product in 38% yield and 98% ee.7a Obviously, the development of Pd-catalyzed asymmetric intermolecular Mizoroki–Heck reaction, particularly, for the enantioselective construction of quaternary carbon is still a formidable challenge.
We have been involved in the program of Pd-catalyzed asymmetric intermolecular Mizoroki–Heck reaction and have realized a switch of regio- and enantio-selectivities in asymmetric Mizoroki–Heck reaction by tuning the electronic properties of bisphosphines and benzylic substituents of ligands respectively.8 These studies and recent progress in this area promoted us to explore the creation of a chiral quaternary carbon center using this reaction. In this communication, we would like to disclose our preliminary results on the Pd-catalyzed asymmetric Heck reaction of 5-substituted-2,3-dihydrofurans with aryl triflates, affording the corresponding Heck products bearing a quaternary carbon center in high yields with excellent enantioselectivity.
We commenced our study with the reaction of 5-methyl-2,3-dihydrofuran (1a) and 4-methoxyphenyl trifluoromethanesulfonate (2a) under the catalysis of Pd(OAc)2 and a commercially available ligand, (R)-BINAP (entry 1, Table 1). The reaction afforded Heck products 3a and 4a in 15% yield (83% ee) and 34% yield (72% ee) respectively. Although these results are promising, further optimization of reaction conditions, such as screening the base, increasing or lowering the reaction temperature, did not improve the reaction (not shown in Table). The reaction always suffered a low selectivity of 3a/4a. Another severe problem is the formation of side-products derived from the ring-opening of the furan ring (see ESI†). These concerns promoted us to use other kinds of ligands. Oestreich's9 and Zhou's5 success of using chiral bisphosphine monoxides for intermolecular Heck reaction inspired us to replace (R)-BINAP with (R)-BINAP(O) as the ligand (entry 2, Table 1). Gratifyingly, the reaction was improved significantly, affording the corresponding Heck product 3a with a chiral quaternary carbon with 88% ee and >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity, but the yield was only 26% with some recovered triflate 2a. Further experiments revealed that the problem was tackled by adding an additional equivalent of (R)-BINAP(O) and product 3a was isolated in 64% yield with 87% ee (entry 3, Table 1). These promising results encouraged us to investigate the impact of the reaction parameters on the reaction. The palladium precursor plays a critical role in the reaction. When Pd(OAc)2 was used in place of Pd(dba)2, a significant drop of the yield to 25% for 3a with only 79% ee was observed (entry 4, Table 1) while the use of [Pd(η3-C3H5)Cl]2 heavily retarded the reaction, forming trace amounts of 3a (entry 5, Table 1). The temperature was found to influence the reaction very much. Lowering the reaction temperature from 110 °C to 90 °C resulted in a lower conversion of triflate 2a (entry 6 vs. entry 3, Table 1). The examination of several other chiral bisphosphine monoxides showed that the reaction with (R)-Segphos(O) gave product 3a in high enantioselectivity but very low yield (entry 7, Table 1). A commercially available duphos monoxide L1 demonstrated no catalytic activity (entry 8, Table 1). The investigation of the solvent effect on the reaction revealed that the use of THF afforded 76% yield of 3a in 87% ee (entry 9 vs. entry 3, Table 1) and the yield of 3a was further increased to 83% by using Cy2NMe instead of iPr2NEt as the base (entry 10, Table 1); (for details of the investigation of the solvent and base effect on the reaction, please see ESI†). Further screening of the chiral bisphosphine monoxide ligand revealed that product 3a in 84% yield with 95% ee and 17:1 regioselectivity was obtained when (R)-SDP(O) was used as the ligand (entry 11 vs. entry 10, Table 1). A more bulky ligand (R)-xyl-SDP(O) proved to be less active and gave inferior olefin selectivity than (R)-SDP(O) (entry 12 vs. entry 11, Table 1). The yield of product 3a dropped significantly when the catalyst loading decreased, and also triflate 2a was not fully consumed (entry 13 vs. entry 11, Table 1). Several other commercially available ligands, such as (S)-tert-BuPHOX, Josiphos, and (R,R)-DACH-phenyl Trost' ligand, were examined, nevertheless, no desired product was acquired (not shown in Table).
:1 regioselectivity, but the yield was only 26% with some recovered triflate 2a. Further experiments revealed that the problem was tackled by adding an additional equivalent of (R)-BINAP(O) and product 3a was isolated in 64% yield with 87% ee (entry 3, Table 1). These promising results encouraged us to investigate the impact of the reaction parameters on the reaction. The palladium precursor plays a critical role in the reaction. When Pd(OAc)2 was used in place of Pd(dba)2, a significant drop of the yield to 25% for 3a with only 79% ee was observed (entry 4, Table 1) while the use of [Pd(η3-C3H5)Cl]2 heavily retarded the reaction, forming trace amounts of 3a (entry 5, Table 1). The temperature was found to influence the reaction very much. Lowering the reaction temperature from 110 °C to 90 °C resulted in a lower conversion of triflate 2a (entry 6 vs. entry 3, Table 1). The examination of several other chiral bisphosphine monoxides showed that the reaction with (R)-Segphos(O) gave product 3a in high enantioselectivity but very low yield (entry 7, Table 1). A commercially available duphos monoxide L1 demonstrated no catalytic activity (entry 8, Table 1). The investigation of the solvent effect on the reaction revealed that the use of THF afforded 76% yield of 3a in 87% ee (entry 9 vs. entry 3, Table 1) and the yield of 3a was further increased to 83% by using Cy2NMe instead of iPr2NEt as the base (entry 10, Table 1); (for details of the investigation of the solvent and base effect on the reaction, please see ESI†). Further screening of the chiral bisphosphine monoxide ligand revealed that product 3a in 84% yield with 95% ee and 17:1 regioselectivity was obtained when (R)-SDP(O) was used as the ligand (entry 11 vs. entry 10, Table 1). A more bulky ligand (R)-xyl-SDP(O) proved to be less active and gave inferior olefin selectivity than (R)-SDP(O) (entry 12 vs. entry 11, Table 1). The yield of product 3a dropped significantly when the catalyst loading decreased, and also triflate 2a was not fully consumed (entry 13 vs. entry 11, Table 1). Several other commercially available ligands, such as (S)-tert-BuPHOX, Josiphos, and (R,R)-DACH-phenyl Trost' ligand, were examined, nevertheless, no desired product was acquired (not shown in Table).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | Base | Solvent | 3a/4ab | Yieldc (3a, %) | eed (%) |
|
a Conditions: molar ratio of 1a/2a/Pd(dba)2/L/base = 500:100:6:12:300.
b Determined by GC.
c Isolated yield.
d Determined by chiral HPLC; a minus sign means that the product has the opposite configuration.
e
1a/2a/Pd(dba)2/L = 500:100:6:6.6.
f Pd(OAc)2 used as the palladium source.
g [Pd(η3-C3H5)Cl]2 used as the palladium source.
h Reaction temperature: 90 °C.
i Performed in a sealed tube due to the low boiling point of THF and high reaction temperature.
j
1a/2a/Pd(dba)2/L = 500:100:3:6.
k ND = not determined.
|
||||||
| 1f | (R)-BINAP | iPr2NEt | THF | 1/2.3 | 15 | −83 |
| 2e | (R)-BINAP(O) | iPr2NEt | 1,4-Dioxane | >99/1 | 26 | −88 |
| 3 | (R)-BINAP(O) | iPr2NEt | 1,4-Dioxane | >99/1 | 64 | −87 |
| 4f | (R)-BINAP(O) | iPr2NEt | 1,4-Dioxane | 5/1 | 25 | −79 |
| 5g | (R)-BINAP(O) | iPr2NEt | 1,4-Dioxane | NDk | Trace | NDk |
| 6h | (R)-BINAP(O) | iPr2NEt | 1,4-Dioxane | >99/1 | 50 | −88 |
| 7 | (R)-Segphos(O) | iPr2NEt | 1,4-Dioxane | NDk | 34 | −85 |
| 8 | L1 | iPr2NEt | 1,4-Dioxane | NDk | Trace | NDk |
| 9i | (R)-BINAP(O) | iPr2NEt | THF | 21/1 | 76 | −87 |
| 10i | (R)-BINAP(O) | Cy2NMe | THF | 65/1 | 83 | −87 |
| 11i | (R)-SDP(O) | Cy2NMe | THF | 17/1 | 84 | 95 |
| 12i | (R)-xyl-SDP(O) | Cy2NMe | THF | 5/1 | 53 | 95 |
| 13i,j | (R)-SDP(O) | Cy2NMe | THF | 18/1 | 48 | 92 |
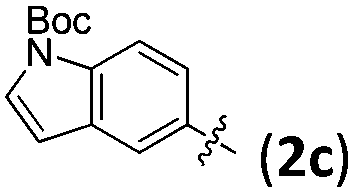
With the optimized conditions in hand, the substrate scope of the reaction was examined and the results are compiled in Table 2. It can be seen that a wide range of dihydrofurans 1 with different substituents, including methyl, propyl, and even bulkier trimethylsilyl, are suitable substrates, affording the corresponding 2,2-disubstituted 2,5-dihydrofurans 3 with a chiral quaternary carbon center in high yield with high regio- and enantioselectivities (entries 1, 11, 13 and 18, Table 2). It is noteworthy that 5-(γ-benzyloxy propyl)-2,3-dihydrofurans 1c is also a suitable substrate (entries 13–17, Table 2). The removal of the benzyloxy group will provide an additional functional group to the product, which makes it more useful in organic synthesis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R (1) | Ar (2) | 3/4b | Yieldc (3, %) | eed (%) |
|
a Conditions: molar ratio of 1/2/Pd(dba)2/(R)-SDP(O)/Cy2NMe = 500:100:6:12:300.
b Determined by GC.
c Isolated yield.
d Determined by chiral HPLC.
e Not determined.
f (R)-BINAP(O) was the ligand; a minus symbol means the opposite sign of optical rotation of the product.
|
|||||
| 1 | Me (1a) | 4-MeO-C6H4 (2a) | 17/1 | 84 (3a) | 95 |
| 2 | 1a | 3,4-(MeO)2-C6H3 (2b) | 32/1 | 96 (3b) | 94 |
| 3 | 1a |
|
21/1 | 74 (3c) | 92 |
| 4 | 1a | 2-Naphthyl (2d) | 17/1 | 57 (3d) | 84 |
| 5 | 1a | Ph (2e) | 25/1 | 63 (3e) | 94 |
| 6 | 1a | 3-Me-C6H4 (2f) | 69/1 | 78f (3f) | −90f |
| 7 | 1a | 2-Me-C6H4 (2g) | 17/1 | 45f (3g) | −96f |
| 8 | 1a | 4-F-C6H4 (2h) | 46/1 | 64 (3h) | 95 |
| 9 | 1a | 3-F-C6H4 (2i) | 22/1 | 31 (3i) | 90 |
| 10 | 1a | 2-F-C6H4 (2j) | 10/1 | 21 (3j) | 90 |
| 11 | n-Pr (1b) | 2a | 8/1 | 57 (3k) | 95 |
| 12 | 1b | 2c | —e | 78 (3l) | 94 |
| 13 | BnO(CH2)3 (1c) | 2a | 59/1 | 83 (3m) | 97 |
| 14 | 1c | 2c | —e | 85 (3n) | 96 |
| 15 | 1c | 2e | 10/1 | 55 (3o) | 93 |
| 16 | 1c | 2f | 9/1 | 63 (3p) | 95 |
| 17 | 1c | 4-CO2Et-C6H4 (2k) | 3.3/1 | 30 (3q) | 70 |
| 18 | SiMe3 (1d) | 2e | 4/1 | 46 (3r) | 93 |
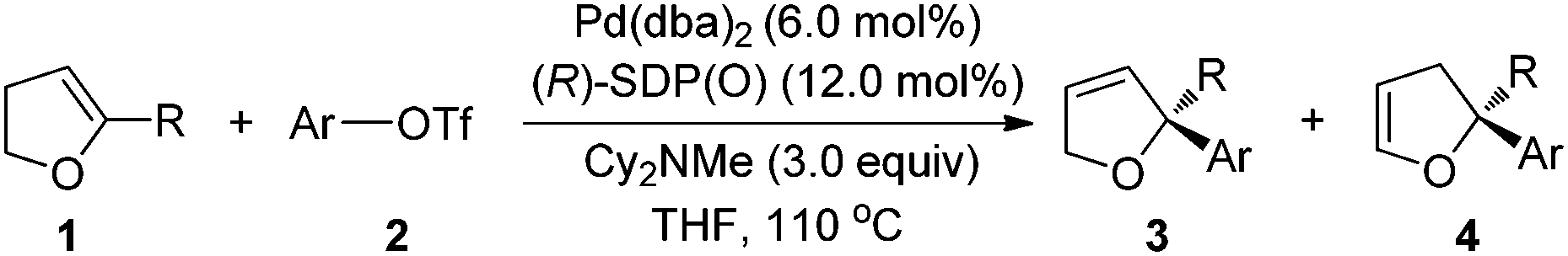
Reaction of different aryl trifluoromethanesulfonates 2 with electron-donating groups at m- and p-position provided the corresponding products in excellent results regarding the yields, regio-, and enantioselectivities (entries 1, 2, 6, 11, 13 and 16, Table 2). Noticeably, heteroaromatic triflate 2c is also a viable substrate to afford desired products in 74–85% yield along with >92% ee (entry 3, 12 and 14, Table 2). In contrast, the Heck products were obtained in low yield for aryl triflates 2 that contain an electron-deficient aromatic ring (entries 9, 10 and 17, Table 2). In these cases, the reduction product of triflates was observed. However, the reaction of 4-fluoro phenyl triflate (2h) afforded rather good results (entry 8, Table 2). It was found that dihydrofuran 3d in 57% yield with 85% ee was obtained by the reaction of 2d with a bulkier naphthyl group (entry 4, Table 2) while the reaction of 2-substituted-aryl triflates 2 afforded lower yields of the corresponding products, also due to the reduction of the triflates (entries 7 and 10, Table 2). Better yields were obtained in the reaction of 1a with 2f and 2g by using (R)-BINAP(O) in place of (R)-SDP(O) as a ligand (entries 6 and 7, Table 2). Some other alkene substrates, such as 5-phenyl-2,3-dihydrofuran, 6-methyl-3,4-dihydro-2H-pyran, and tert-butyl 5-methyl-2,3-dihydro-1H-pyrrole-1-carboxylate, were examined, but the corresponding Heck reaction product was not observed (not shown in Table 2).
This Heck reaction can be scaled up to a 2.0 mmol scale. Treatment of 512 mg of triflate 2a with 672 mg of 5-methyl-2,3-dihydrofuran (1a) under the optimized reaction conditions provided 334 mg of product 3a in 88% yield with 95% ee. The absolute configuration of product 3e was determined to be S by comparing its sign of optical rotation with that reported in the literature.10a
These chiral quaternary carbon-containing Heck reaction products were transformed into other important building blocks easily according to the known methods.10 Allylic oxidation of the Heck reaction products 3a and 3e using PCC in CH2Cl2 in the presence of pyridine, a procedure reported by Hong's group,10a afforded butenolides 5a and 5b10a,14 with a chiral quaternary carbon center, respectively, without the loss of optical activity (eqn (1)), which is an important structural motif presented in many natural products and biologically active compounds such as rugulovasine A11 and (+)-pinnatolide.12 This approach provides a simple and efficient way to synthesize these optically active γ-disubstituted α,β-unsaturated butenolides and represents one of the few catalytic examples for their synthesis.13 The absolute configuration of product 3e was determined as S by comparing the sign of optical rotation of butenolides 5b with that reported in the literature.14 It should be noted that partial racemization occurred in the allylic oxidation of compounds 3a and 3e if chromium trioxide was used as the oxidant according to the procedure of Marco's group,10b,c
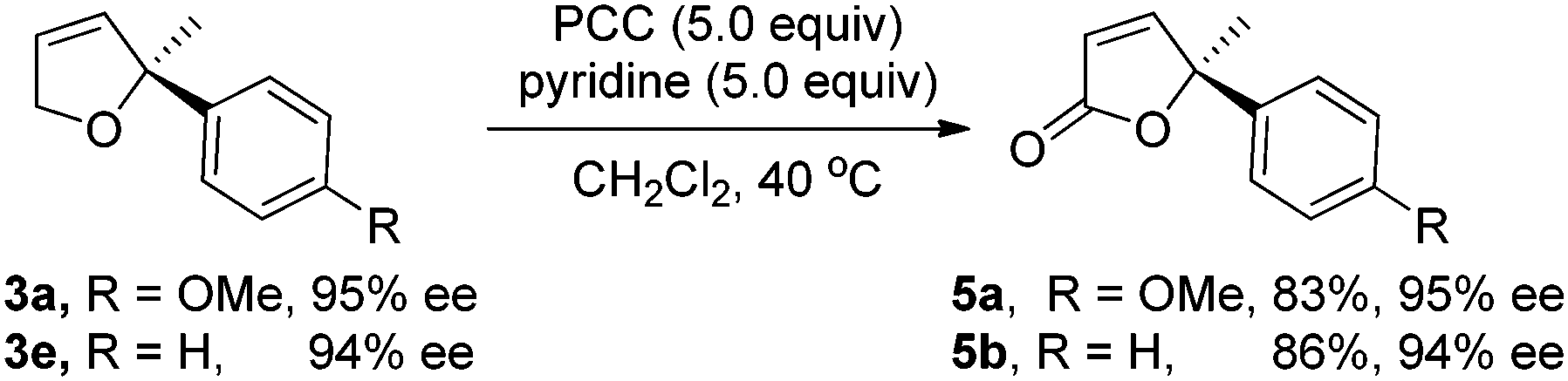 | (1) |
To gain more understanding on the beneficial effect of an additional ligand on the reaction, some controlled experiments were performed with (R)-BINAP(O) as an example due to its more ready availability than (R)-SDP(O). Using 6 mol% of Pd(dba)2 and (R)-BINAP(O) in the presence of an external achiral 1,2-bis(diphenylphosphino)ethane monoxide [DPPE(O)] as the catalyst, product 3a in 64% yield with 86% ee was afforded. It can also be noted that product 3a in 88% ee was obtained if the catalyst with 1:1 ratio of Pd/(R)-BINAP(O) was used (entry 2, Table 1), which means that the additional ligand did not improve the enantioselectivity and it was not involved in the enantioselectivity-determining step of the reaction. All of these facts mean that the additional ligand plays a role in stabilizing the Pd(0) species in order to improve the yield of the reaction.
In conclusion, we have realized a palladium-catalyzed asymmetric intermolecular Mizoroki–Heck reaction for the construction of a chiral quaternary stereocenter, affording 2,2-disubstituted 2,5-dihydrofurans in high yields with excellent enantioselectivity. The importance of the presence of an additional ligand in the reaction has been found, and its role has also been studied.
This work was financially supported by the Major Basic Research Development Program (2011CB808706), National Natural Science Foundation of China (NSFC) (21372242, 21472214, and 21421091), the NSFC and the Research Grants Council of Hong Kong Joint Research Scheme (21361162001), the Chinese Academy of Sciences, the Technology Commission of Shanghai Municipality, and the Croucher Foundation of Hong Kong.
Notes and references
- (a) A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1994, 33, 2379 CrossRef PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (c) The Mizoroki–Heck Reactions, ed. M. Oestreich, John Wiley & Sons, New York, 2009 Search PubMed.
- F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417 CrossRef CAS.
- For selected reviews, see: (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS PubMed; (c) L. F. Tietze, H. Ila and H. P. Bell, Chem. Rev., 2004, 104, 3453 CrossRef CAS PubMed; (d) D. McCartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (e) M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282 CrossRef CAS PubMed ; For selected examples, see: ; (f) W. J. Drury, N. Zimmermann, M. Keenan, M. Hayashi, S. Kaiser, R. Goddard and A. Pfaltz, Angew. Chem., Int. Ed., 2004, 43, 70 CrossRef PubMed; (g) J. Mazuela, O. Pàmies and M. Diéguez, Chem. – Eur. J., 2010, 16, 3434 CrossRef CAS PubMed; (h) M. O. Fitzpatrick, H. Muller-Bunz and P. J. Guiry, Eur. J. Org. Chem., 2009, 1889 CrossRef CAS PubMed; (i) S. T. Henriksen, P.-O. Norrby, P. Kaukoranta and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 10414 CrossRef CAS PubMed; (j) C. C. Oliveira, A. G. Salles, E. A. F. Santos and C. R. D. Correia, Tetrahedron Lett., 2012, 53, 3325 CrossRef PubMed.
- (a) K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neill and K. W. Jung, Org. Lett., 2007, 9, 3933 CrossRef CAS PubMed; (b) S. Sakaguchi, K. S. Yoo, J. O'Neill, J. H. Lee, T. Stewart and K. W. Jung, Angew. Chem., Int. Ed., 2008, 47, 9326 CrossRef CAS PubMed; (c) K. S. Yoo, J. O'Neill, S. Sakaguchi, J. H. Lee and K. W. Jung, J. Org. Chem., 2010, 75, 95 CrossRef CAS PubMed.
- (a) Y. Zhang and J. R. Zhou, J. Am. Chem. Soc., 2012, 134, 11833 CrossRef PubMed; (b) J. Hu, H. R. Hirao, Y. X. Li and J. R. Zhou, Angew. Chem., Int. Ed., 2013, 52, 8676 CrossRef CAS PubMed; (c) J. Hu, Y. P. Lu, Y. X. Li and J. R. Zhou, Chem. Commun., 2013, 49, 9425 RSC; (d) S. J. Liu and J. R. Zhou, Chem. Commun., 2013, 49, 11758 RSC; (e) C. L. Wu and J. R. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS PubMed.
- (a) E. W. Werner, T. S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (b) T. S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) C. C. Oliveira, R. A. Angnes and C. R. D. Correia, J. Org. Chem., 2013, 78, 4373 CrossRef CAS PubMed; (d) Y. Dang, S. Qu, Z.-X. Wang and X. Wang, J. Am. Chem. Soc., 2014, 136, 986 CrossRef CAS PubMed; (e) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960 CrossRef CAS PubMed; (f) T. S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed.
- (a) M. Tschoerner and P. S. Pregosin, Organometallics, 1999, 18, 670 CrossRef CAS ; Formation of a chiral quaternary carbon center using substrate-induced strategy: ; (b) P. Nilsson, M. Larhed and A. Hallberg, J. Am. Chem. Soc., 2003, 125, 3430 CrossRef CAS PubMed.
- (a) T. Tu, W. P. Deng, X. L. Hou, L. X. Dai and X. C. Dong, Chem. – Eur. J., 2003, 9, 3073 CrossRef CAS PubMed; (b) X. L. Hou, D. X. Dong and K. Yuan, Tetrahedron: Asymmetry, 2004, 15, 2189 CrossRef CAS PubMed; (c) W.-Q. Wu, Q. Peng, D.-X. Dong, X.-L. Hou and Y.-D. Wu, J. Am. Chem. Soc., 2008, 130, 9717 CrossRef CAS PubMed; (d) C.-H. Ding and X.-L. Hou, Bull. Chem. Soc. Jpn., 2010, 83, 992 CrossRef CAS; (e) H. Li, C.-H. Ding, B. Xu and X.-L. Hou, Acta Chim. Sin., 2014, 72, 765 CrossRef CAS.
- (a) T. H. Wöste and M. Oestreich, Chem. – Eur. J., 2011, 17, 11914 CrossRef PubMed; (b) T. H. Wöste and M. Oestreich, ChemCatChem, 2012, 4, 2096 CrossRef PubMed ; For other applications of bisphosphine monoxides in catalysis, see: ; (c) A. B. Charette, A. Côté, J. N. Desrosiers, I. Bonnaventure, V. N. G. Lindsay, C. Lauzon, J. Tannous and A. A. Boezio, Pure Appl. Chem., 2008, 80, 881 CrossRef CAS; (d) V. V. Grushin, Chem. Rev., 2004, 104, 1629 CrossRef CAS PubMed; (e) M. McConville, O. Saidi, J. Blacker and J. Xiao, J. Org. Chem., 2009, 74, 2692 CrossRef CAS PubMed.
- (a) Y. Wang, K. Zheng and R. Hong, J. Am. Chem. Soc., 2012, 134, 4096 CrossRef CAS PubMed; (b) M. Carda, E. Castillo, S. Rodríguez, S. Uriel and J. A. Marco, Synlett, 1999, 1639 CrossRef CAS PubMed; (c) J. A. Marco, M. Carda, S. Rodríguez, E. Castillo and M. N. Kneeteman, Tetrahedron, 2003, 59, 4085 CrossRef CAS.
- (a) M. Abe, S. Ohmono, T. Ohashi and T. Tabuchi, Agric. Biol. Chem., 1969, 33, 469 CrossRef CAS; (b) R. J. Cole, J. W. Kirksey, J. Clardy, N. Eickman, S. M. Weinreb, P. Singh and D. Kim, Tetrahedron Lett., 1976, 17, 3849 CrossRef.
- F. Bohlmann, C. Zdero and L. Lehmann, Phytochemistry, 1991, 30, 1161 CrossRef.
- (a) A. Quintard, A. Lefranc and A. Alexakis, Org. Lett., 2011, 13, 1540 CrossRef CAS PubMed; (b) C. K. Tan, J. C. Er and Y.-Y. Yeung, Tetrahedron Lett., 2014, 55, 1243 CrossRef CAS PubMed.
- (a) K. Suzuki and K. Inomata, Tetrahedron Lett., 2003, 44, 745 CrossRef CAS; (b) C. Gaul, K. Schärer and D. Seebach, J. Org. Chem., 2001, 66, 3059 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc03601a |
| This journal is © The Royal Society of Chemistry 2015 |