
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface plasmon & visible light for polymer functionalization of mesopores and manipulation of ionic permselectivity†
Nicole
Herzog
a,
Jonas
Kind
a,
Christian
Hess
b and
Annette
Andrieu-Brunsen
*a
aTechnische Universität Darmstadt, Ernst-Berl-Institut für Technische und Makromolekulare Chemie, Alarich-Weiss-Str. 4, D-64287 Darmstadt, Germany. E-mail: brunsen@cellulose.tu-darmstadt.de
bTechnische Universität Darmstadt, Eduard-Zintl-Institut für Anorganische und Physikalische Chemie, Alarich-Weiss-Str. 8, D-64287 Darmstadt, Germany
First published on 16th June 2015
Abstract
The near-field of surface plasmons is locally confined to nanometer dimensions. Here, we use surface plasmons to initiate polymer-functionalization of mesoporous silica films allowing subsequent ionic permselectivity gating based on polymer charge. We expect this functionalization approach to open a new dimension of functional miniaturization e.g. in nanofluidics.
Many efforts are concentrated on the fabrication of miniaturized structures and building of nanodevices. To miniaturize devices and functionalization the limits of synthetic protocols have to be adapted to confine synthesis to the nanoscale. Spatiotemporal control over a polymerization reaction is mainly offered by photo-initiation procedures.1 Recently, even sunlight was proposed to be used for a controlled polymerization.2 Using conventional photolithography, local resolution is limited by Abbe's law of diffraction.3 On the other hand, the electromagnetic field of near-field modes is characterized by local nanometer dimensions and tunable wavelength in the visible. Surprisingly, surface plasmons are mainly used for sensing4,5 and only few studies on surface-plasmon induced polymerization are reported.6,7 Polymerization in the visible can for example, be achieved with multicomponent initiating systems, such as the combination of dyes as photo-sensitizer and amines as radical initiating molecules.8 Examples are cyanine dyes,9,10 methylene blue,11 or alexa dyes.12–14 Combining such visible-light photo-initiation concepts with mesoporous membranes (surface) functionalization of mesopores, visible light-initiated, localized near-field functionalization in water becomes accessible.15 Very recently the group of Soppera6,16 as well as the group of Kreiter7,17 demonstrated the possibility to initiate a solution photopolymerization with the surface plasmon of metallic nanoparticles. In this context Chegel and co-workers used surface plasmon at a planar metal interface to perform photochemistry and to in situ generate a biosensor matrix without taking care about surface immobilization.11 So far these experiments are focused on generating a relatively undefined polymer matrix and they are not taking into account the third dimension perpendicular to a surface or covalent surface grafting of the generated polymer. Besides, polymer-functionalized mesoporous thin films are known to be able to gate ionic permselectivity by excluding ions of identical charge and pre-concentrating countercharged ions.18,19 Such functionalized membranes are highly interesting for nanofluidics, filtration-sensing elements, sensor arrays.20
The applied concept of visible light induced surface polymer functionalization of mesoporous thin silica films is summarized in Fig. 1.
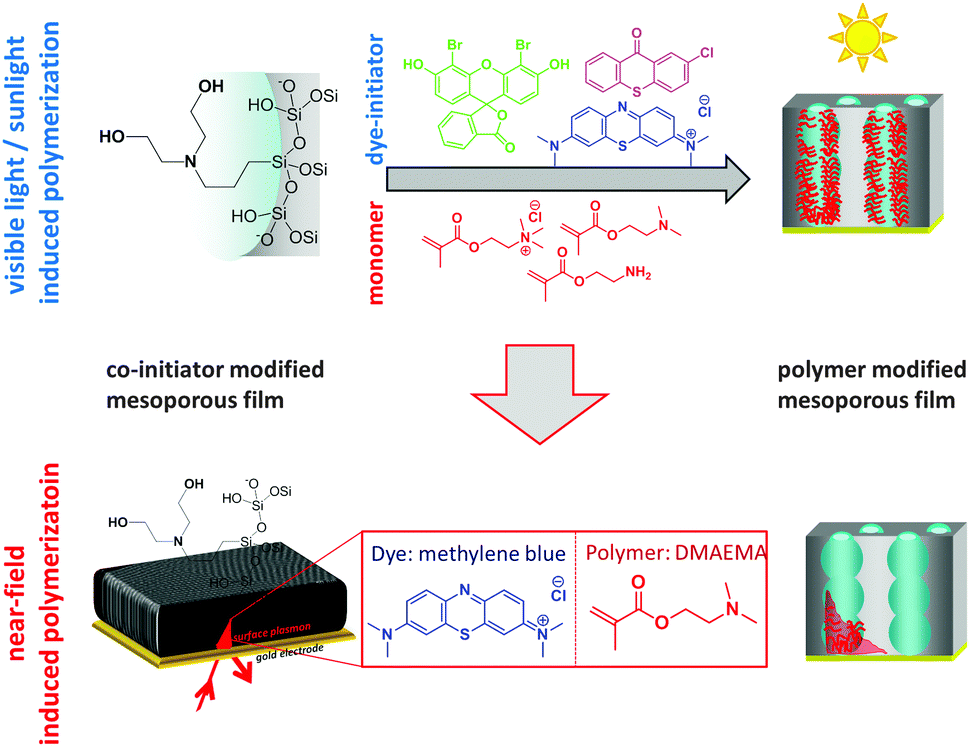 | ||
| Fig. 1 Schematic presentation of the visible light, sunlight, or surface plasmon near-field induced surface polymer modification of mesoporous thin silica films using dye-sensitized polymerizations. | ||
Three different dyes with absorption wavelengths of 390 nm (2-chlorothioxanthone), 510 nm (4′,5′-dibromofluorescein), and 660 nm (methylene blue) in combination with bis-(2-hydroxyethyl)-3-aminopropyltriethoxysilane as co-initiator6,21 are used for visible light initiated photopolymerization. The absorption spectra (Fig. 2) clearly show that the absorption maxima are well separated, covering a wavelength range from 400 to almost 700 nm. This allows to individually address three different wavelengths for synthetic modifications.
 | ||
| Fig. 2 UV-vis spectra of the applied dye initiators 2-chlorothioxanthone (orange), 4′,5′-dibromofluorescein (green), and methylene blue (blue) measured in a water-based solution. | ||
To ensure covalent polymer attachment to the mesopore wall we covalently grafted the co-initiator 3-[bis(2-hydroxyethyl)amino]propyl-triethoxysilane to the mesoporous silica thin films. After silica surface modification with the co-initiator, XPS analysis (Fig. 3) revealed one co-initiator molecule per approximately ten silica atoms in case of functionalization at an elevated temperature of 80 °C for 225 minutes. Reducing the reaction time to 45 minutes resulted in a slightly decreased co-initiator concentration of one molecule per 12.5 silica atoms (Fig. S3, ESI†) as extracted from integration of the N1s signals at around 400 eV.
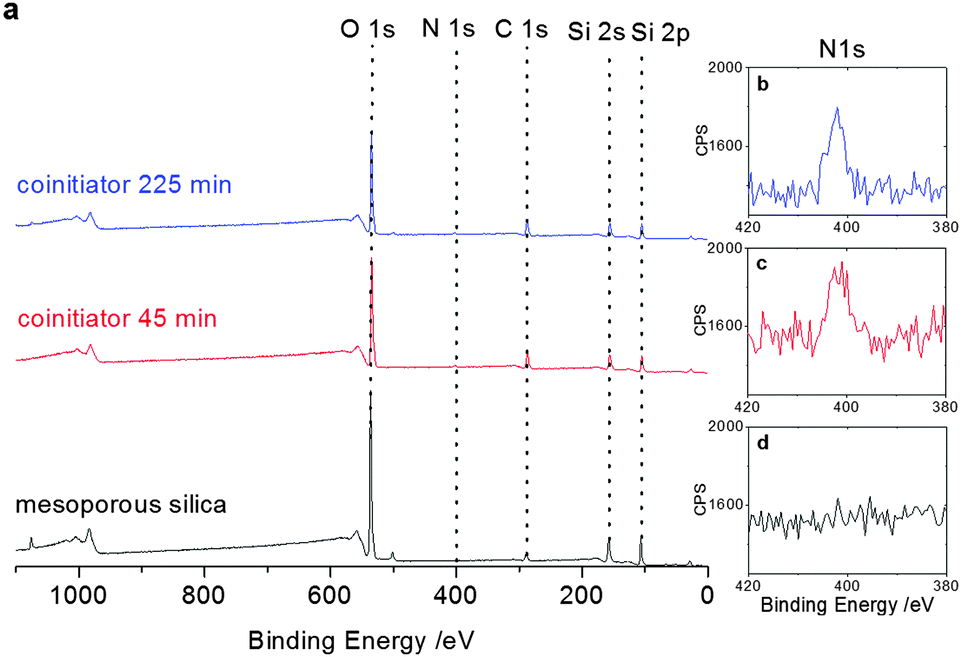 | ||
| Fig. 3 (a) XP-spectra showing the co-initiator presence after functionalization for 45 minutes (red) and 225 minutes (blue) in comparison to non-functionalized mesoporous thin films (black) including the respective zoom into the N1s signal for the untreated mesoporous silica film (d) as well as after functionalization for 45 minutes (c) and 225 minutes (b). | ||
Subsequently, the mesoporous silica films were functionalized with polymer using 2-aminoethylmethacrylate (AEMA), 2-(dimethylamino)ethylmethacrylate (DMAEMA) or [2-(methacryloyloxy)ethyl]trimethylammonium chloride (METAC) by irradiation with visible light, sunlight or surface-plasmon near field modes in the presence of the initiating dye. Experimental details can be found in the ESI.†
Initially, polymer functionalization using an external irradiation source in the presence of different monomers and dyes is discussed. Polymer functionalization is monitored by following the polymer C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O infrared absorption at 1725 cm−1 (Fig. 4a). All infrared spectra show the characteristic bands of the inorganic framework and the organic functions incorporated into the mesoporous films after extraction of unbound monomer and polymer.22 All spectra are normalized to the silica matrix using the transverse optical Si–O–Si modes at 1065 cm−1 (νas). Especially, the remaining CO stretching at 1725 cm−1 after extensive extraction indicates the successful visible-light induced polymerization at the mesoporous silica thin film. To proof that polymerization occurs as well inside the mesopores and not primarily on the external film surface, a short CO2 plasma treatment was used to destroy the co-initiator located at the external mesoporous film surface.22 Subsequent polymerization as well shows polymer functionalization with only slightly reduced CO infrared absorption at 1725 cm−1 (Fig. 4b) indicating the polymer functionalization of the mesopore walls.
O infrared absorption at 1725 cm−1 (Fig. 4a). All infrared spectra show the characteristic bands of the inorganic framework and the organic functions incorporated into the mesoporous films after extraction of unbound monomer and polymer.22 All spectra are normalized to the silica matrix using the transverse optical Si–O–Si modes at 1065 cm−1 (νas). Especially, the remaining CO stretching at 1725 cm−1 after extensive extraction indicates the successful visible-light induced polymerization at the mesoporous silica thin film. To proof that polymerization occurs as well inside the mesopores and not primarily on the external film surface, a short CO2 plasma treatment was used to destroy the co-initiator located at the external mesoporous film surface.22 Subsequent polymerization as well shows polymer functionalization with only slightly reduced CO infrared absorption at 1725 cm−1 (Fig. 4b) indicating the polymer functionalization of the mesopore walls.
 | ||
| Fig. 4 Infrared spectra showing the generation of polymer as detected by the CO vibration at 1725 cm−1 for the polymerization of (a) METAC with methylene blue (red), DMAEMA with 4′,5′-dibromofluorescein (cyan), methylene blue (blue), 2-chlorothioxanthone (marine) and AEMA with 4′,5′-dibromofluorescein (olive), methylene blue (dark yellow), 2-chlorothioxanthone (green) for non-plasma treated samples and (b) mesoporous silica films after plasma treatment and thus destruction of the co-initiator at the external mesoporous film surface. (c) Schematic view of the near-field induced polymerization in the Kretschmann configuration. The angular spectra of a ∼260 nm (red) and a ∼550 nm (blue) thick mesoporous film are shown in the presence of air (d) and monomer solution (f). (e) Infrared spectra for the polymer PDMAEMA (red) and the PDMAEMA modified mesoporous ∼260 nm film (black) in comparison to the DMAEMA monomer (grey). In case of reduced laser intensity, shorter reaction time and an angle of almost no energy coupling (64°) in case of the ∼550 nm film only a very low amount of polymer is generated (g, blue). | ||
Comparing the IR absorption at 1725 cm−1 (CO) for METAC and AEMA with plasma treatment (Fig. 4b) and without plasma treatment (Fig. 4a) a slightly reduced absorption mostly between 10–20% and up to 50% in case of METAC polymerization with methylene blue is observed. This indicates a reduction in polymer amount relative to the silica amount for plasma-treated films as compared to non-plasma treated samples. The observed reduction in CO absorption is not proportional to the inner versus outer surface ratio which might indicate diffusional limitations for the polymerization in mesopores under the applied experimental conditions. Nevertheless, a significant amount of polymer is generated within the mesopores (Fig. 4b) showing the diffusion of dye and monomer into the mesoporous film and the potential of visible light induced polymerization as functionalization strategy for the inner mesopore surface. Polymerization inside the mesopores was additionally verified by ellipsometry measurements. The film thickness was determined to be around 260 nm after polymerization and the refractive index increased upon each modification step from 1.26 to 1.36 after co-initiator modification, exceeding 1.40 up to 1.46 after polymerization. Based on the effective medium approximation23 a total pore filling of 30–45% in case of METAC polymerization initiated with methylene blue is detected. Analyzing the solution polymer for of DMAEMA polymerization by GPC measurements, a Mw between 1 × 105 and 7 × 105 with a PDI between 2.7 and 5.7 was observed after initiation with 4′,5′-dibromofluorescein or 2-chlorothioxanthone. In case of polymerization at planar silicon wafer substrates the solution polymer molecular weight according to GPC measurements was smaller (3–8 × 104 g mol−1). The exact values are summarized in Table S2 (ESI†). For surface polymer analysis an exemplary degrafting for PDMAEMA initiated with methylene blue and with 2-chlorothioxanthone revealed very small amounts of polymer and molecular weights of 5 monomer units per chain as determined by mass spectroscopy. This supports earlier observations on polymerization in confinement of mesopores resulting in lower polymer molecular weight as compared to polymerization at planar surfaces and in solution.22,24 Further details can be found in the ESI.†
Ellipsometry, infrared spectroscopy, and GPC analysis of the solution polymer indicate a successful polymerization initiated by visible light between 400 and 700 nm for three different initiators and three different monomers METAC, AEMA, DMAEMA. This is building the basis for localized polymerization initiated by near field modes such as surface plasmons. Even sunlight exposure resulted in polymerization as verified by infrared spectroscopy. In the following, the above discussed polymerization conditions are transferred to surface plasmon-initiated surface polymer functionalization of a mesoporous silica film supported on a thin gold film.
Under the applied experimental conditions surface plasmons are induced between 400–700 nm in the Kretschmann configuration (Fig. 4c). Surface plasmons generated in a 50 nm gold film with a He–Ne-laser excitation at 633 nm in the Kretschmann configuration25 are characterized by an exponentially decaying electromagnetic field perpendicular to the metal surface with a penetration depth of ∼200 nm.25 We functionalized mesoporous silica films by using the highly localized surface plasmon electromagnetic field to initiate dye-sensitized polymerization at 633 nm. At this wavelength methylene blue as initiating dye is used.
Mesoporous silica thin films were deposited on a gold-covered (50 nm) high refractive index LaSFN9 glass substrate. The gold layer is covered with a few nm thick, sputtered SiOx layer to facilitate mesoporous film deposition by dip coating and evaporation induced self-assembly (EISA).26 Irradiation with a He–Ne-laser in the Kretschmann configuration results in generation of a surface plasmon under resonant angle (Fig. 4d and f). The resonant angle for the mesoporous silica film deposited on the SiOx-covered gold interface is around 60–65° in the presence of air and shifts to up to 75° upon addition of water-based monomer solution (Fig. 4d and f). The mesoporous film thickness was determined to be around 260 nm for a one layer mesoporous film and around 550 nm for a two layer mesoporous film, which is in accordance with ellipsometry measured on wafer substrates. Surface plasmon-induced polymerization was carried out at the surface plasmon resonance angle with a laser irradiation input energy of up to 4 mW and constant flow of monomer/dye solution at a flow rate of 5 μl min−1. Taking into account the loss of energy, visible by the coupling of the surface plasmon, 35% of the irradiated energy in case of the one layer film (Fig. 4d red) is transmitted into the surface plasmon and thus used for polymerization initiation. After dismantling the flow cell, polymer generation for a thin, 260 nm thick, film was already visible by eye proving the potential of surface plasmons for polymerization initiation at functionalization of interfaces. Polymer generation was as well supported by infrared spectroscopy clearly showing polymer generation by the disappearing double bond signals at 2770 cm−1 and 1640 cm−1 (Fig. 4e) comparing monomer and polymer infrared spectra. Additionally, the presence of polymer vibrational bands in the absence of monomer vibrational bands for the surface-plasmon induced polymerization in a mesoporous silica film (Fig. 4e) supports polymer functionalization. Performing the surface plasmon-initiated polymerization with a thicker ∼560 nm mesoporous silica film at an angle of 64°, shorter reaction time and under reduced irradiation energy, no or only very small DMAEMA signals were visible in the infrared spectrum (Fig. 4g). This clearly shows the great potential of near-field induced polymerizations for gradual functionalization of mesoporous films applicable in ionic permselectivity control, as well as the huge potential for nanoscale local control on polymer functionalization by benefiting from nanoscale near-field local distribution.
The applicability of visible-light polymer functionalization of mesoporous films to control ionic permselectivity is demonstrated by cyclic voltammetry measurements (Fig. 5). The exemplarily shown cyclic voltammogramms for unmodified mesoporous silica (Fig. 5a) and poly-DMAEMA modified mesoporous silica after plasma treatment and 4′,5′-dibromofluorescein initiated polymerization (CO absorption 0.05 at 1725 cm−1) (Fig. 5b) clearly show the influence of polymer functionalization within the mesopores. Due to the positively charged poly-DMAEMA the mesoporous film becomes anion-selective and excludes positively charged probe molecules at an acidic pH, whereas the negatively charged molecules are able to enter the pores to a higher concentration as compared to unmodified mesoporous silica films. This is demonstrated by the higher peak current in case of poly-DMAEMA modified films (Fig. 4b) as compared to unmodified silica mesoporous films (Fig. 4a). More results on ionic permselectivity can be found in the ESI.†
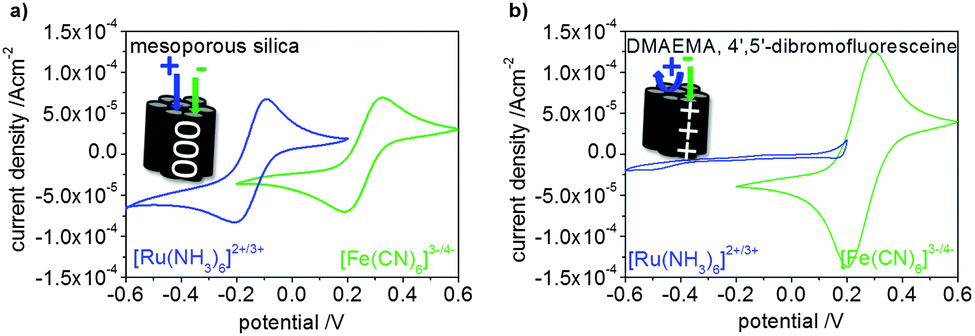 | ||
| Fig. 5 Cyclic voltammetry (25 mV s−1) results at an acidic pH (2–3) clearly show the influence of the polymer (here DMAEMA) on ionic permselectivity (b) for a positively charged probe molecule ([Ru(NH3)6]2+/3+ blue) and a negatively charged probe molecule ([Fe(CN)6]3−/4− green) in comparison to unmodified mesoporous silica (a). | ||
In conclusion, we have developed a mesopore polymer functionalization strategy based on visible light induced radical polymerization which allows mesopore polymer functionalization by near-field modes. Our results suggest polymer functionalization of the inner mesopore walls with lower molecular weights as compared to the polymer generated in solution. Polymer functionalization results in ion permselective behavior governed by the polymer charge. This mesopore functionalization approach is expected to have great potential for local synthetic control with resolution on the nanometer scale and thus especially in the field of nanofluidics and lab on chip devices.
The authors would like to thank the Landesoffensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz (LOEWE Soft Control) as well as the Fonds der chemischen Industrie and the Adolf-Messer Stiftung for recognition and financial support of this work. The authors thank Karl Kopp for performing the XPS experiments and Hamid Keshmiri for initial work on mesoporous film preparation on gold surfaces.
References
- Z. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schöder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- Z. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290 CrossRef CAS PubMed.
- W. Knoll, A. Kasry, F. Yu, Y. Wang, A. Brunsen and J. Dostalek, J. Nonlinear Opt. Phys. Mater., 2008, 17, 121–129 CrossRef CAS.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386–406 RSC.
- C. Deeb, C. Ecoffet, R. Bachelot, J. Plain, A. Bouhelier and O. Soppera, J. Am. Chem. Soc., 2011, 133, 10535–10542 CrossRef CAS PubMed.
- M. Alvarez, A. Best, A. Unger, J. M. Alonso, A. del Campo, M. Schmelzeisen, K. Koynov and M. Kreiter, Adv. Funct. Mater., 2010, 20, 4265–4272 CrossRef CAS PubMed.
- J.-P. Fouassier, F. Morlet-Savary, J. Lalevée, X. Allonas and C. Ley, Materials, 2010, 3, 5130–5142 CrossRef CAS PubMed.
- J. Paczkowski and J. Kabatc, Dyes Pigm., 2010, 86, 133–142 CrossRef PubMed.
- O. Valdes-Aguilera, C. P. Pathak, J. Shi, D. Watson and D. C. Neckers, Macromolecules, 1992, 25, 541–547 CrossRef CAS.
- V. Chegel, M. J. Whitcombe, N. W. Turner and S. A. Piletsky, Biosens. Bioelectron., 2009, 24, 1270–1275 CrossRef CAS PubMed.
- J. M. Belisle, J. P. Correia, P. W. Wiseman, T. E. Kennedy and S. Costantino, Lab Chip, 2008, 8, 2164–2167 RSC.
- J. M. Belisle, D. Kunik and S. Costantino, Lab Chip, 2009, 9, 3580–3585 RSC.
- M. A. Holden and P. S. Cremer, J. Am. Chem. Soc., 2003, 125, 8074–8075 CrossRef CAS PubMed.
- J. C. Scaiano and K. G. Stamplecoskie, J. Phys. Chem. Lett., 2013, 4, 1177–1187 CrossRef CAS.
- H. Ibn-El-Ahrach, R. Bachelot, G. Lerondel, A. Vial, A. S. Grimault, J. Plain, P. Royer and O. Soppera, J. Microsc., 2008, 229, 421–427 CrossRef CAS PubMed.
- K. H. Dostert, M. Alvarez, K. Koynov, A. del Campo, H. J. Butt and M. Kreiter, Langmuir, 2012, 28, 3699–3703 CrossRef CAS PubMed.
- G. J. A. A. Soler-Illia and O. Azzaroni, Chem. Soc. Rev., 2011, 40, 1107–1150 RSC.
- S. Alberti, G. J. A. A. Soler-Illia and O. Azzaroni, Chem. Commun., 2015, 51, 6050–6075 RSC.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC.
- I. Dika, J.-P. Malval, O. Soppera, V. Bardinal, D. Barat, C. Turck, A. Spangenberg and A. Bruyant, Chem. Phys. Lett., 2011, 515, 91–95 CrossRef CAS PubMed.
- L. Silies, H. Didzoleit, C. Hess, B. Stühn and A. Andrieu-Brunsen, Chem. Mater., 2015, 27, 1971–1981 CrossRef CAS.
- J. E. Spanier and I. P. Herman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 10437 CrossRef CAS.
- A. Andrieu-Brunsen, S. Micoureau, M. Tagliazucchi, I. Szleifer, O. Azzaroni and G. J. A. A. Soler-Illia, Chem. Mater., 2015, 27, 808–821 CrossRef CAS.
- W. Knoll, Annu. Rev. Phys. Chem., 1998, 49, 569–638 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc03507d |
| This journal is © The Royal Society of Chemistry 2015 |