
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free hydrogen evolution with nanoparticles derived from pyrene via two-photon ionization induced by laser irradiation†
Kei
Ohkubo
*ab,
Naoki
Kohno
a,
Yusuke
Yamada
ac and
Shunichi
Fukuzumi
*abd
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: ookubo@chem.eng.osaka-u.ac.jp; fukuzumi@chem.eng.osaka-u.ac.jp
bDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
cDepartment of Applied Chemistry & Bioengineering, Graduate School of Engineering, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
dFaculty of Science and Technology, Meijo University and ALCA and SENTAN, Japan Science and Technology Agency (JST), Tempaku, Nagoya, Aichi 468-8502, Japan
First published on 9th June 2015
Abstract
Laser irradiation of a cyclohexane solution containing pyrene resulted in hydrogen evolution as pyrene was converted to a metal-free nanoparticle photocatalyst. When C6H12 was replaced by C6D12, D2 was mainly evolved. This result suggests that the hydrogen source is cyclohexane used as a solvent. Photocatalytic hydrogen evolution was also observed in an aqueous solution by using a water-soluble pyrene derivative.
Various approaches for light energy conversion into chemical energy have been developed in artificial photosynthesis.1–4 Heterogeneous metallic nanoparticles composed of semiconductor metallic oxides have been widely used for water splitting as catalysts in hydrogen production from water.5–8 However, the catalytic mechanism of hydrogen evolution is yet to be clarified. On the other hand, molecular metal-free photocatalysis under homogeneous conditions has recently gained special attention in organic synthesis, physical chemistry and green chemistry because of its low cost and mild reaction conditions for activation of substrates.9–13 Preparation of organic nanocrystals has been established by laser induced decomposition of organic crystals and particles.14–18 However, there have been few reports on photocatalytic reactions using heterogeneous organic nanoparticles as metal-free photocatalysts.19,20
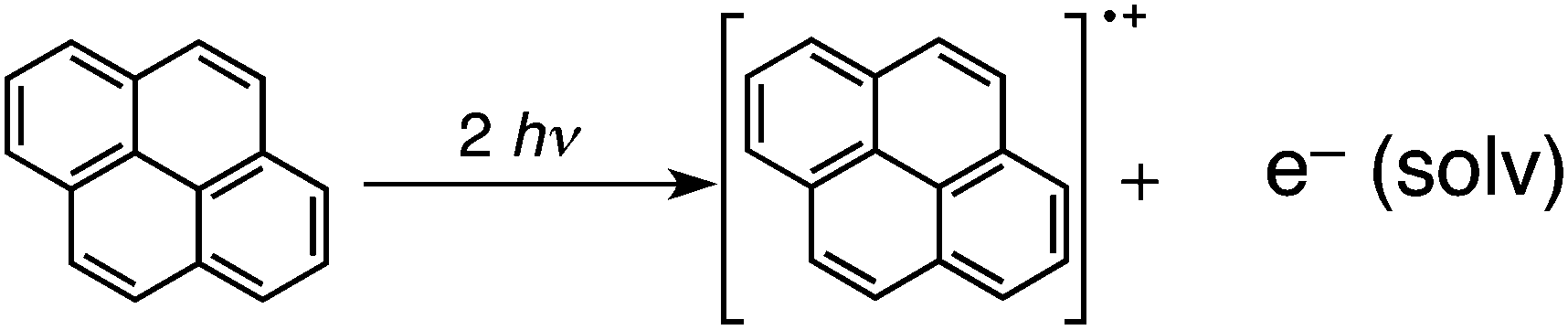
Pyrene is known as an organic photosensitiser in the photochemical reactions. Two-photon excitation of pyrene produces photoionization products, the pyrene radical cation and a solvated electron under homogeneous conditions. The one-electron oxidation and reduction potential of pyrene is +1.3 and −2.0 V vs. SCE in MeCN.21,22 The pyrene radical anion is generated by two-photon excitation and one-electron reduction by the solvated electron [e−(solv)] or photoinduced electron transfer between the excited state of a pyrene molecule (S2 or S1 state) and the ground state of another pyrene molecule due to the higher-lying excited state (E(S1) = 3.34 eV)23–26 than the HOMO–LUMO gap (3.30 eV) in polar solvent. In nonpolar solvent, the solvated electron may have strong reducing ability to reduce a proton (H+) to hydrogen (H2) without a metal catalyst.
H2 is primarily used in the chemical industry as a reactant and is proposed as an alternative energy source for the future. Catalytic H2 evolution systems have extensively been studied by using sacrificial electron donors, photosensitisers and electron mediators such as methyl viologen and an H2 evolution catalyst such as platinum.27–32 However, there has been no report on photocatalytic H2 evolution systems via proton reduction using an organic photosensitiser alone without any sacrificial electron source.
We report herein that metal-free photoinduced H2 evolution has been made possible by using pyrene as a precursor for nanoparticles. Efficient H2 evolution was observed in various organic solvents as well as water using pyrene alone as a metal-free organic photocatalyst under laser light irradiation at room temperature under atmospheric pressure. The reaction mechanism of laser-induced H2 evolution has been clarified based on the oxidized products, deuterium kinetic isotope effects and the dependence of the rate of H2 evolution on the laser intensity.
Nanosecond laser flash irradiation (λ = 355 nm, 10 Hz, 40 mJ pulse−1, i.d. 8 mm) of a deaerated cyclohexane solution containing pyrene (50 mM) for 90 min resulted in the formation of highly dispersed black nanoparticles in the solution. Fig. 1 shows the UV-vis absorption spectral change upon laser irradiation of pyrene in cyclohexane. The characteristic absorption bands at 306, 319 and 334 nm due to pyrene observed before irradiation disappeared and turned to the broad absorption from the UV-vis to near-IR region.
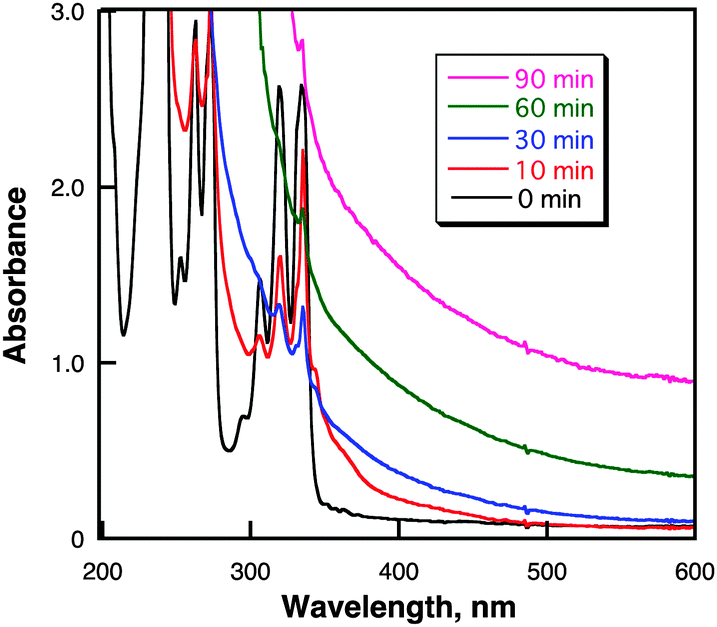 | ||
| Fig. 1 UV-vis absorption spectral changes for growing pyrene nanoparticles observed in a cyclohexane solution (2.5 mL) containing pyrene (50 μM) under laser irradiation (λ = 355 nm; 40 mJ pulse−1; 10 pulse s−1). | ||
To characterize the nanoparticles, dynamic light scattering (DLS) measurements were performed for the photochemically growing nanoparticles. The nanoparticles of 420 nm size were observed after laser irradiation for 30 min as shown in Fig. 2a.
 | ||
| Fig. 2 Time course of DLS data of a cyclohexane solution (2.5 mL) containing pyrene (50 μM) under laser irradiation (λ = 355 nm; 40 mJ pulse−1; 10 Hz). | ||
The particle size grows by further laser irradiation to afford nanoparticles of 1100 nm size after the irradiation time of 90 min. Transmission electron microscopy (TEM) measurements were performed to evaluate the transformation of pyrene nanoparticles after laser irradiation. TEM images after photoirradiation for 30 min (Fig. 3a and b) clearly exhibit small nanoparticles. After irradiation for 90 min, the size of nanoparticles was enlarged (Fig. 3c and d). The IR spectra of nanoparticles showed that the characteristic peak at 3043 cm−1 due to the C–H vibration disappeared and it was blue-shifted to 2925 cm−1 with an increase in irradiation time (see Fig. S1 in the ESI†). This suggests that C–H bonds of pyrene were broken in growing nanoparticles. To obtain the information of the C–H bond of pyrene in the nanoparticle, CHN elemental analysis was performed in growing nanoparticles. The ratio of carbon and hydrogen was 94.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.6, which is virtually the same as the comparison of pyrene alone (95.0:5.0). Thus, the main component in the nanoparticle is the undamaged pyrene. The pyrene molecules on the particle surface may be dehydrogenated to form a pyrene polymer by the C–C bond coupling reaction. The formation of pyrene oligomers was confirmed by the powder X-ray diffraction (PXRD) measurements (Fig. S2 in the ESI†). The XRD patterns with peaks at 2θ = 11 and 24° due to the (001) and (220) planes were maintained after laser irradiation, indicating that the pyrene was not damaged in the nanoparticles.
:5.6, which is virtually the same as the comparison of pyrene alone (95.0:5.0). Thus, the main component in the nanoparticle is the undamaged pyrene. The pyrene molecules on the particle surface may be dehydrogenated to form a pyrene polymer by the C–C bond coupling reaction. The formation of pyrene oligomers was confirmed by the powder X-ray diffraction (PXRD) measurements (Fig. S2 in the ESI†). The XRD patterns with peaks at 2θ = 11 and 24° due to the (001) and (220) planes were maintained after laser irradiation, indicating that the pyrene was not damaged in the nanoparticles.
 | ||
| Fig. 3 TEM images of (a) a cyclohexane solution (2.5 mL) of pyrene (50 μM) after laser light irradiation (λ = 355 nm; 40 mJ pulse−1; 10 pulse s−1) for 30 min, (b) magnified figure of (a), (c) dispersion liquid of precipitation after laser light irradiation (λ = 355 nm; 40 mJ pulse−1; 10 pulse s−1) for 90 min and (d) magnified figure of (c). | ||
Laser induced nanoparticle formation was initiated by the photo-ionization of pyrene to form a pyrene radical cation and a solvated electron [eqn (1)]. Hydrogen atom transfer from the cyclohexane radical anion (Cy˙−), which is produced by the reduction of cyclohexane by solvated electron [eqn (2)], to the pyrene radical cation (Py˙+) occurs to produce a hydrogenated pyrene neutral radical (PyH˙) and a cyclohexyl radical (C6H11˙) [eqn (3)]. Two molecules of PyH˙ react to give the pyrene dimer and H2 [eqn (4)]. The dimer was detected by MALD I-TOF-MS
 | (1) |
| C6H12 + e−(solv) → C6H12˙− | (2) |
| Py˙+ + C6H12˙− → PyH˙ + C6H11˙ | (3) |
| 2PyH˙ → Py–Py + H2 | (4) |
On the basis of the above-mentioned reaction mechanism in eqn (4), the laser-induced dimerization of pyrene radicals results in H2 evolution. H2 evolution was successfully detected by GC experiments in the reaction of pyrene in cyclohexane under nanosecond laser irradiation at 355 nm as shown in Fig. 4. The TON of the catalysts for H2 evolution was 2000 for 7 h based on the initial moles of pyrene in cyclohexane where the reaction conditions are [Py] = 50 μM, 40 mJ pulse−1 at λ = 355 nm in cyclohexane (2.5 mL). No deactivation was observed in laser irradiation for 7 h (Fig. S5 in the ESI†). When cyclohexane was replaced by cyclohexane-d12 as a solvent, D2 gas was selectively evolved (data are shown in Fig. S6 in the ESI†).33 The kinetic isotope effect (KIE) value for H2 evolution was determined to be 2.1. Thus, the rate-determining step of H2 evolution is carbon–hydrogen bond cleavage of the cyclohexane radical anion by hydrogen atom transfer to the pyrene radical cation. Indeed, no H2 evolution was observed in cyclohexene instead of cyclohexane (Fig. S7 in the ESI†). H2 evolution also occurred when the laser light excitation wavelength was changed from 355 nm to 532 nm by an Nd:YAG laser as a light source (Fig. S8 in the ESI†) because pyrene nanoparticles show a strong and broad absorption band covering the whole visible region as previously shown in Fig. 1.
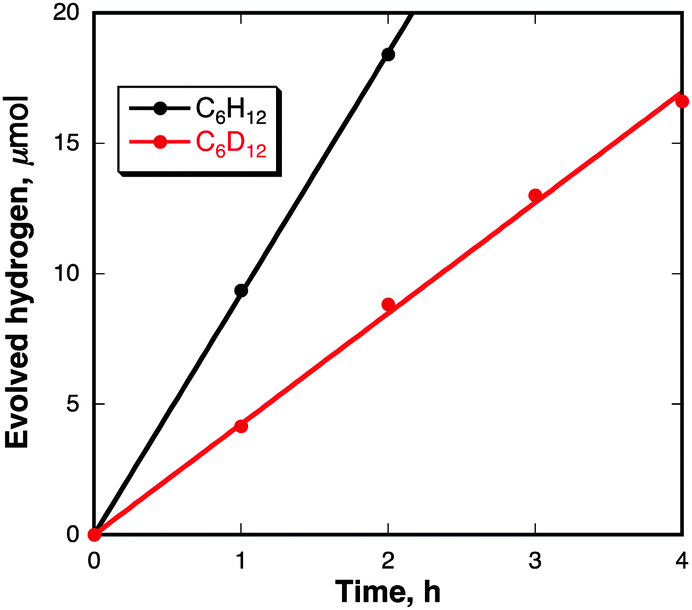 | ||
| Fig. 4 Time courses of the amount of evolved H2 and D2 under laser irradiation (λ = 355 nm; 20 mJ pulse−1; 10 pulse s−1) of a cyclohexane or cyclohexane-d12 solution (2.5 mL) containing pyrene (0.5 mM). | ||
The dependence of the rate of H2 evolution on the laser intensity was examined using different laser power intensities at 355 nm (0–20 mJ pulse−1) as shown in Fig. 5a. The initial rates of H2 evolution are proportional to the second power of the laser intensity as shown in Fig. 5b. This suggests that an ionization reaction of two-photon absorbed species may be involved in the photocatalytic H2 evolution. The maximum quantum yield observed was 7.9% when the laser power is 40 mJ pulse−1 at 355 nm. The efficiency of the photocatalytic H2 evolution is affected by the solvent polarities as shown in Table 1. The efficiency of photocatalytic H2 evolution is highest for cyclohexane and lowest for dimethylsulfoxide in the series of solvents.34 This suggests that electron reducing ability of solvated electron generated by photoionisation of pyrene in non-polar cyclohexane is much higher than in the polar solvent due to the stabilization of solvated electron.
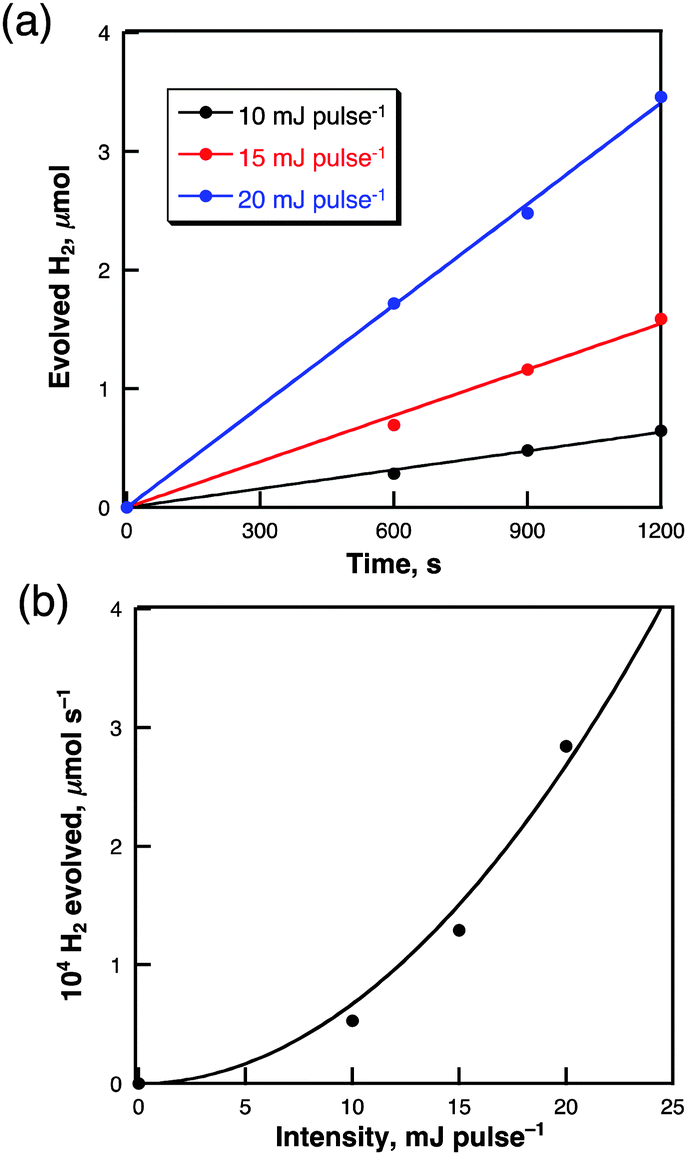 | ||
| Fig. 5 (a) Time courses of the amount of evolved H2 in cyclohexane (2.5 mL) containing pyrene particles (0.25 mg) under laser light irradiation (λ = 355 nm; 10, 15 or 20 mJ pulse−1; 10 pulse s−1) and (b) Plot of the rate of H2 evolution vs. laser intensity. A fitted line was drawn by a second-order function. | ||
| Solvent | Dielectric constant | H2 (μmol h−1) |
|---|---|---|
| a Conditions: solvent: 2.5 mL; pyrene: 0.50 mM; nanosecond laser (λ = 355 nm; 20 mJ pulse−1; 10 pulse s−1), under N2. | ||
| n-Heptane | 1.9 | 5.7 |
| Cyclohexane | 2.0 | 14 |
| 2-Propanol | 18 | 2.6 |
| Acetone | 20 | 3.2 |
| Acetonitrile | 37 | 4.6 |
| Dimethylsulfoxide | 47 | 0.1 |
On the basis of the above-mentioned results, the reaction mechanism of photocatalytic H2 evolution from pyrene nanoparticles (Py-NP) in cyclohexane (CyH) is shown in eqn (5)–(9). Two-photon ionization of Py-NP is much easier than the pyrene monomer, yielding a Py-NP radical cation (Py-NP˙+) and a cyclohexane radical anion (C6H12˙−) [eqn (5)]. Hydrogen atom transfer from C6H12˙− to Py-NP˙+ occurs [eqn (6)], followed by electron transfer from C6H11− to Py-NP(–H)+ [eqn (7)] and radical coupling with the release of H2 [eqn (8)]. Cyclohexyl radicals (C6H11˙) split disproportionately to yield cyclohexene and cyclohexane [eqn (9)]. The total stoichiometry is given by eqn (10) by summing up eqn (5)–(9).
| Py-NP + C6H12 + 2hν → Py-NP˙+ + C6H12˙− | (5) |
| Py-NP˙+ + C6H12˙− → Py-NP(–H)+ + C6H11− | (6) |
| Py-NP(–H)+ + C6H11− → Py-NP(–H)˙ + C6H11˙ | (7) |
| 2Py-NP(–H)˙ → (Py-NP)2 + H2 | (8) |
| 2C6H11˙ → C6H10 + C6H12 | (9) |
| nPy-NP + (n − 1)C6H12 → (Py-NP)n + (n − 1)C6H10 + (n − 1)H2 | (10) |
No nanoparticle of pyrene was formed in an aqueous solution by the laser photoirradiation at 355 nm. Thus, no H2 evolution was observed under photoirradiation. When pyrene was replaced by sodium pyrene-1-acetate used as a water-soluble photosensitiser, the nanoparticle formation and H2 evolution occurred in an aqueous solution under otherwise the same reaction conditions (Fig. S9 in the ESI†). We also examined H2 evolution using other hydrocarbons instead of pyrene. Nanoparticles were also formed under the same laser-irradiation conditions. Nanoparticles of coronene and 9,10-diphenylanthracene were effective as H2-evolution photocatalysts. However, the rates of H2 evolution were 1.4, 1.1 and 0 μmol h−1 for 9,10-diphenylanthracene, coronene and rubrene, respectively, which are significantly lower than the value of pyrene (4.6 μmol h−1) in acetonitrile. In the case of rubrene as a red dye, no nanoparticle was formed without H2 evolution. It is difficult to form oligomers as precursors of H2-evolution catalysts because of the more delocalized π-radical cation than pyrene radical cation. In particular, the radical cations of rubrene and 9,10-diphenylanthracene are very stable in solution without any reaction such as dimerization.
In conclusion, pyrene nanoparticles have been demonstrated to act as efficient metal free organic photocatalysts for H2 evolution from various solvents. Cyclohexane used as a solvent is the most effective hydrogen source in H2 evolution with pyrene nanoparticles. The rate of H2 evolution increased with increasing laser intensity, exhibiting a second power dependence, because H2 was evolved via the two-photon ionization of pyrene followed by hydrogen atom transfer from the cyclohexane radical anion to the pyrene radical cation. Pyrene radical cations dimerise to form oligomer nanoparticles as more effective photocatalysts for H2 evolution. This is the first report of catalytic H2 evolution using an organic nanoparticle photocatalyst without inorganic material.
This work was supported by Grants-in-Aid (no. 26620154 and 26288037 to K.O. and no. 24350069 and 25600025 to Y.Y.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); ALCA and SENTAN projects from JST, Japan (to S.F.). We acknowledge Research Centre for Ultra-Precision Science & Technology in Osaka University for TEM measurements.
Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC
.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- M. Hambourger, A. Brune, D. Gust, A. L. Moore and T. A. Moore, Photochem. Photobiol., 2005, 81, 1015–1020 CrossRef CAS PubMed
- R. Abe, K. Hara, K. Sayama, K. Domen and H. Arakawa, J. Photochem. Photobiol., A, 2000, 137, 63–69 CrossRef CAS
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed
-
(a) P. D. Morse and D. A. Nicewicz, Chem. Sci., 2015, 6, 270–274 RSC
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
- A. Aguirre-Soto, C.-H. Lim, A. T. Hwang, C. B. Musgrave and J. W. Stansbury, J. Am. Chem. Soc., 2014, 136, 7418–7427 CrossRef CAS PubMed
-
(a) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed
- E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017–2037 CAS
-
(a) T. Asahi, T. Sugiyama and H. Masuhara, Acc. Chem. Res., 2008, 41, 1790–1798 CrossRef CAS PubMed
- H. Tabata, M. Akamatsu, M. Fujii and S. Hayashi, Jpn. J. Appl. Phys., 2007, 46, 4338–4343 CrossRef CAS
- A. Ibanez, S. Maximov, A. Guiu, C. Chaillout and P. L. Baldeck, Adv. Mater., 1998, 10, 1540–1543 CrossRef CAS
- J. Ye, H.-Z. Chen and M. Wang, J. Mater. Sci., 2003, 38, 4021–4025 CrossRef CAS
- A. H. Matsui, K. Mizuno, O. Nishi, Y. Matsushima, M. Shimizu, T. Goto and M. Takeshima, Chem. Phys., 1995, 194, 167–174 CrossRef CAS
-
(a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- K. Ohkubo, N. Kohno, Y. Yamada and S. Fukuzumi, Chem. Sci., 2015, 6, 666–674 RSC
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem. – Eur. J., 2003, 9, 1585–1593 CrossRef CAS PubMed
- S. Fukuzumi, J. Yuasa, N. Satoh and T. Suenobu, J. Am. Chem. Soc., 2004, 126, 7585–7594 CrossRef CAS PubMed
- J. T. Richards, G. West and K. T. John, J. Phys. Chem., 1970, 74, 4137 CrossRef
- A. R. Watkins, J. Phys. Chem., 1976, 80, 713–717 CrossRef CAS
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6470 CrossRef CAS PubMed
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1972 CrossRef CAS PubMed
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- R. M. Navarro, M. C. A. Galván, J. A. V. de la Mano, S. M. Al-Zahrani and J. L. G. Fierro, Energy Environ. Sci., 2010, 3, 1865–1882 CAS
- H. Zhou, T. Fan and D. Zhang, ChemSusChem, 2011, 3, 513–528 CAS
-
(a) K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed
-
(a) H. Kotani, K. Ohkubo, Y. Takai and S. Fukuzumi, J. Phys. Chem. B, 2006, 110, 24047–24053 CrossRef CAS PubMed
- HD was formed by the detection with GC as shown in Fig. S6a in the ESI.† HD is a coupling product in the reaction of PyD˙ and PyH˙ [eqn (4)].
- Lower H2 formation efficiency in more non-polar n-heptane than cyclohexane may result from the lower numbers of weak secondary C–H bonds in n-heptane.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic details. See DOI: 10.1039/c5cc03501e |
| This journal is © The Royal Society of Chemistry 2015 |