
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombination of inverse electron-demand Diels–Alder reaction with highly efficient oxime ligation expands the toolbox of site-selective peptide conjugations†
S.
Hörner‡
a,
C.
Uth‡
a,
O.
Avrutina
a,
H.
Frauendorf
b,
M.
Wiessler
c and
H.
Kolmar
*a
aTechnische Universität Darmstadt, Clemens-Schöpf-Institut für Organische Chemie und Biochemie, Alarich-Weiss Straße 4, 64287 Darmstadt, Germany. E-mail: kolmar@biochemie-tud.de
bGeorg-August-Universität Göttingen, Institut für Organische und Biomolekulare Chemie, Zentrale Analytik/Massenspektrometrie, Tammannstraße 2, 37077 Göttingen, Germany
cDeutsches Krebsforschungszentrum, Medizinische Physik in der Radiologie, Projektgruppe Biologische Chemie E020, Im Neuenheimer Feld, 69120 Heidelberg, Germany
First published on 10th June 2015
Abstract
A modular approach combining inverse electron-demand Diels–Alder coupling (DARinv) and oxime ligation expands the toolbox of bioorthogonal peptide chemistry. Applicability of versatile site-specific bifunctional building blocks is demonstrated by generation of defined conjugates comprising linear, cystine-bridged and multi-disulfide functional peptides as well as their conjugation with hybrid silsesquioxane nanoparticles.
Covalent linkage of certain peptidic units, being an alternative or a complementary strategy to recombinant production, often becomes a method of choice for the synthesis of sophisticated macromolecular constructs with tailored properties.1 Indeed, to date a vast number of bioconjugates and the respective techniques have been reported,2 ranging from relatively simple fluorescently3 or small-molecule-labelled peptides4 to complex, multifunctional architectures like antibody–drug conjugates.5 Obviously, chemical transformations suitable for bioconjugations must satisfy at least two obligatory requirements, chemoselectivity and efficiency.6 In view of the variety of inherent functional groups present in peptidic molecules, the development of a viable orthogonal chemistry for their effective junction at a certain position still remains a challenge.
Generally, the strategy towards site-specific bioconjugations7 relies on incorporation of a uniquely addressable group at the desired position in the molecule of interest followed by its peculiar reaction with the respective counterpart. Such a uniquely addressable moiety could be incorporated into peptidic molecules through a vast number of post-synthetic modifications, e.g. periodate oxidation of β-aminoalcohols,8,9 or via the non-natural building blocks10 either upon recombinant production11 or in the course of chemical synthesis. Bioorthogonal reactions to target these non-natural functional groups often make use of rich ketone and aldehyde chemistry12 as well as numerous click-type reactions,3,13 with the azide–alkyne cycloaddition being the most prominent representative.14–16 During the last decade, a special class of pericyclic reactions has got the highest priority as they utilize the ring strain to promote increased reactivity upon cycloaddition.17–19 In particular, the Diels–Alder reaction with inverse electron-demand (DARinv)20–22 between numerous dienophiles23–25 and tetrazines21,26,27 was found to be a valuable tool for effective bioorthogonal conjugations.28–30 Followed by a retro-Diels–Alder reaction to eliminate nitrogen gas, this so-called tetrazine ligation is characterized by extremely fast kinetics with second-order rate constants up to 2 × 103 M−1 s−1 and has been already used in a number of both in situ and in vivo studies.25,31,32
In this study we present a modular approach to the conjugation of biomolecules based on the combination of two efficient chemical transformations, oxime ligation and DARinv. For the proof-of-concept experiments we designed bifunctional building blocks to incorporate the bioorthogonal DARinv coupling site in a peptide site-specifically via oximation of the respective aldehyde. Following this step, attachment of a DARinv counterpart would accomplish a desired conjugate. The choice of the strategy was specified by two arguments, feasible generation of required aldehydes in biomolecules8,9,33 and fast kinetics of an irreversible DARinv. We reasoned that our approach, initially investigated on peptides of different size and molecular complexity, could be further extended to orthogonal conjugations featuring a broad spectrum of biomacromolecules, i.e. proteins, sugars, or other biopolymers.
Here it is important to mention that, while having been used for numerous labeling approaches,34,35 the DARinv reaction was surprisingly rarely applied to connect functional peptides site-specifically.28,36 This could be explained taking into account that both the diene and the dienophile counterparts have been to date installed into partner biomolecules using either amide37,38 or maleimide chemistry39 which cannot provide the required orthogonality, especially with regard to cysteine/cystine-bearing molecules. Though the installation of DARinv building blocks into peptide-like molecules on solid support has been reported, this approach did not find broad application. Moreover, the on-resin assembly is very rarely used for the production of full-size proteins.
Since oxime ligation has been applied to couple exo-norbornene as a dienophile handle onto aldehydes of the reducing ends of oligosaccharides40 and taking into consideration that this moiety is easily generated in both recombinant and synthetic peptides41 without interference with functional side chains, we decided to use this highly efficient reaction to decorate the peptides of interest with the respective diene and dienophile partners.
Our modular approach relies on the tailor-made bifunctional building blocks equipped with an aminooxy moiety for the primary incorporation into peptidic counterparts along with the DARinv site for the successive ligation (Fig. 1). In our proof-of-concept study we installed the DARinv building blocks 5 and 8 into the next functional peptides: a Sortase A recognition heptapeptide 12,42,43 an integrin-binding RGD decapeptide 9,12 an antimicrobial decapeptide Jelleine derivative 13,44 a disulfide-bridged pentadecapeptide matriptase inhibitor 10,45 and two cystine knots comprising three disulfides and a backbone of more than thirty amino acids 11, 1446 (Scheme S2, ESI†). An additional N-terminal serine was introduced into each peptide to provide the orthogonally addressable site upon post-synthetic modification.9 The resulted glyoxylyl moieties generated by periodate oxidation (Scheme S1, ESI†) were oximated by the aminooxy-bearing derivatives of Reppe anhydride 5 and tetrazine 8, respectively (Fig. 1 and 2). The dienes 15–17 were synthesized from peptide-glyoxals 9–11, and the dienophiles 18–20 – from peptide-glyoxals 12–14 (Fig. 2 and ESI 1.2.12†). Peptides were assembled by microwave-assisted Fmoc-SPPS.47 Following cleavage from the support with successive oxidative folding if required, the N-terminal serines were oxidized by NaIO4 to generate the aminooxy-reactive glyoxals 9–14 for further conjugations (ESI 1.2.9–1.2.11†).
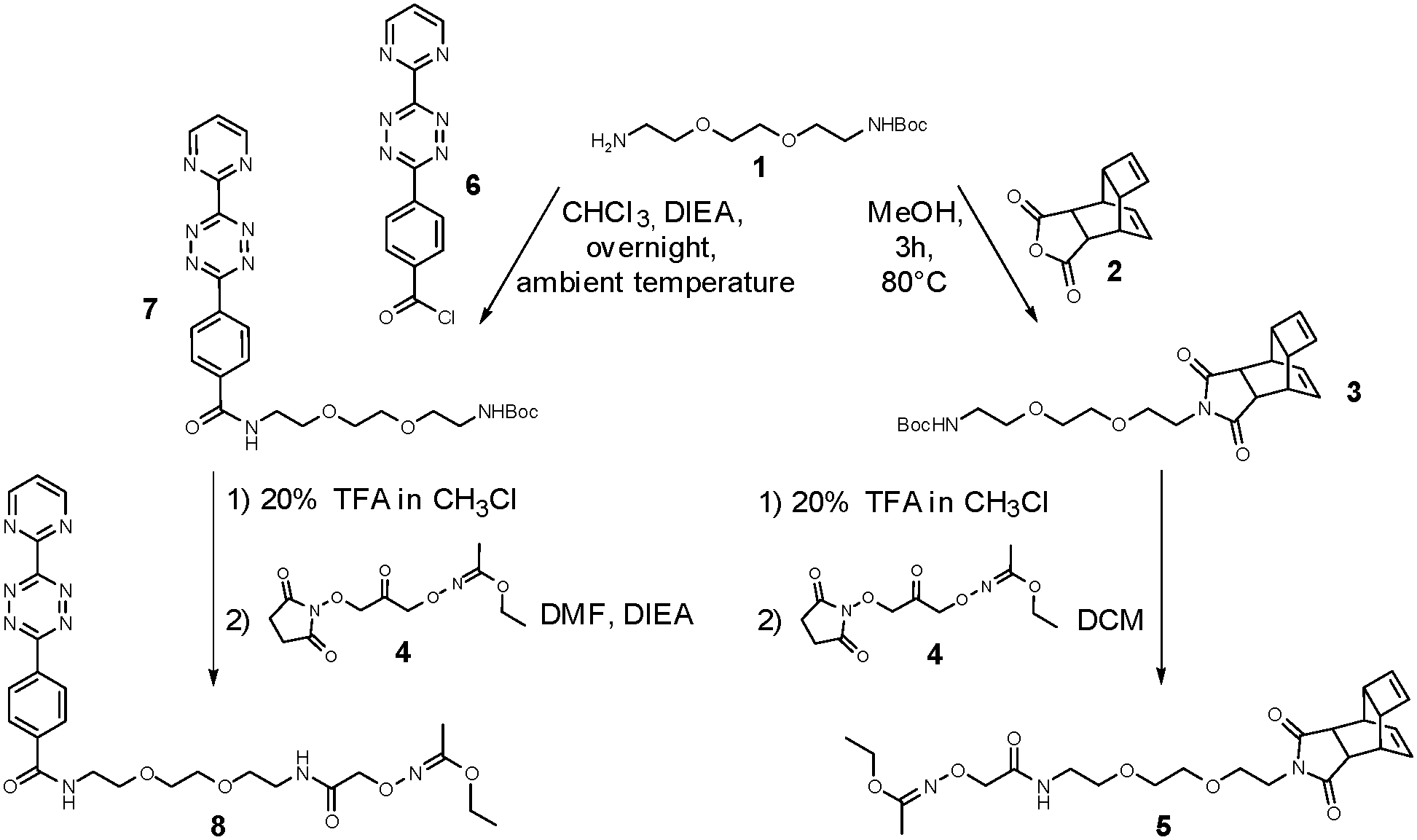 | ||
| Fig. 1 Synthesis of DARinv building blocks. | ||
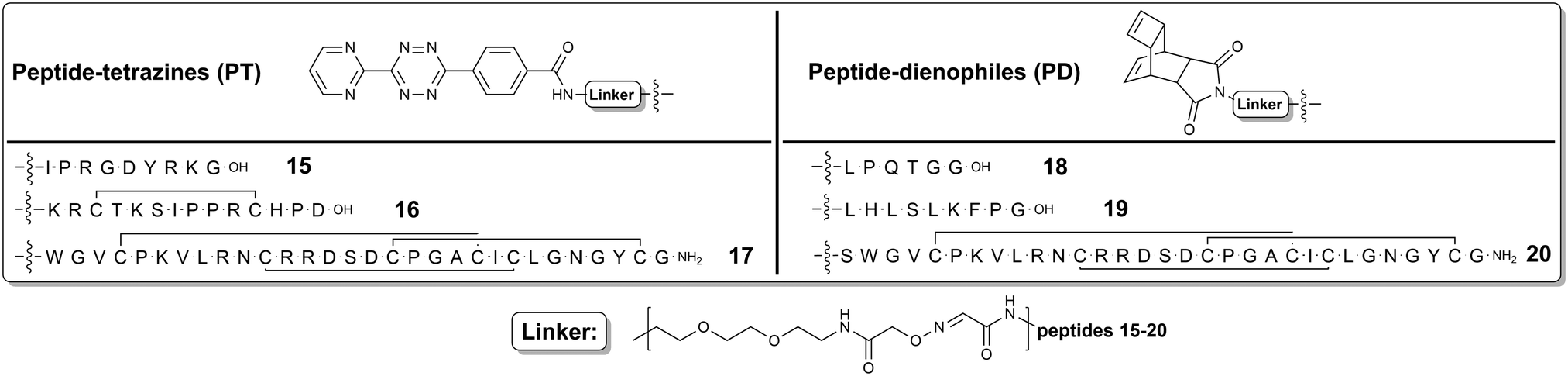 | ||
| Fig. 2 Synthesized peptidic ligands for successive DARinv conjugations. | ||
Prior to the installation into synthetic peptides, the bifunctional DARinv building blocks were synthesized following a two-step procedure as illustrated in Fig. 1.
Thus, mono-Boc-protected miniPEG 1 was reacted with Reppe anhydride 2 or tetrazine derivative 6, respectively, and the resulting constructs 3 and 7 were transformed into N-ethoxyethylidene (Eei)-protected aminooxy building blocks 5 and 8 upon acidolytic cleavage of N-Boc protection followed by the N-acylation with N-hydroxysuccinimide-activated (NHS) Eei-protected aminooxy acetic acid 4 (Fig. 1 and ESI 1.2.1–1.2.8†).48 The oxime ligation was performed in 50% (v:v) aqueous TFA overnight leading to peptide-tetrazines 15–17 and peptide-dienophiles 18–20 (Fig. 2 and ESI 1.2.12†).
After the conjugation partners have been decorated with the respective DARinv building blocks, the resulted diene and dienophile counterparts were reacted with each other in 10% aq. acetonitrile containing 0.1% TFA overnight at ambient temperature giving conjugates 21–29 (Fig. 3 and 4, ESI 1.2.15†). The exemplified monitoring of DARinv reaction progress presented in Fig. 4 and in ESI 1.2.15† clearly indicates that conversion into the desired products has been achieved for all constructs. Interestingly, upon evaluation of the mass spectrometric data, we observed for some DARinv conjugates the recovery of the aromatic system in the tetrazine, as previously reported.49,50 The mass spectra can be seen in Fig. 5 as well as in ESI 1.2.21–1.2.29.†
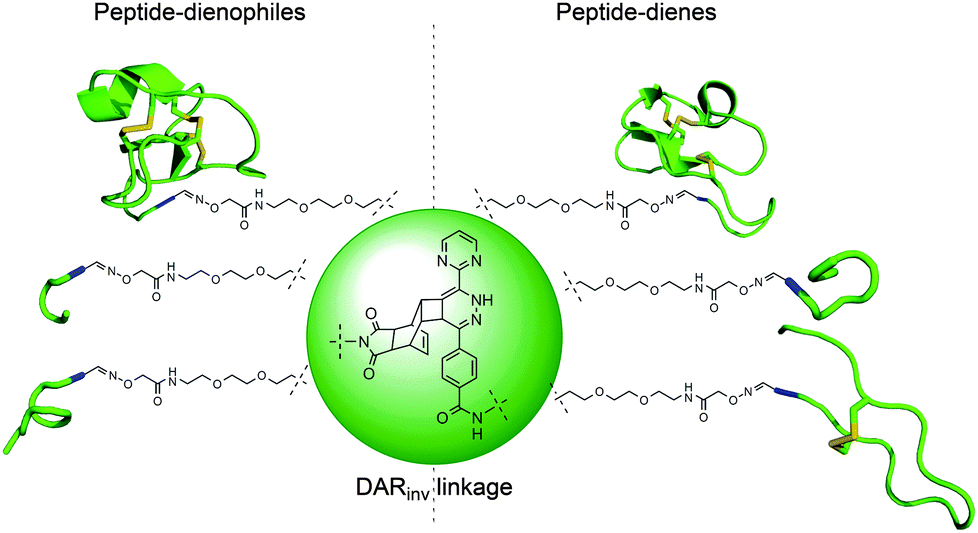 | ||
| Fig. 3 Schematic representation of peptide conjugates synthesized by DARinv between the counterparts depicted on the left and the right panels. | ||
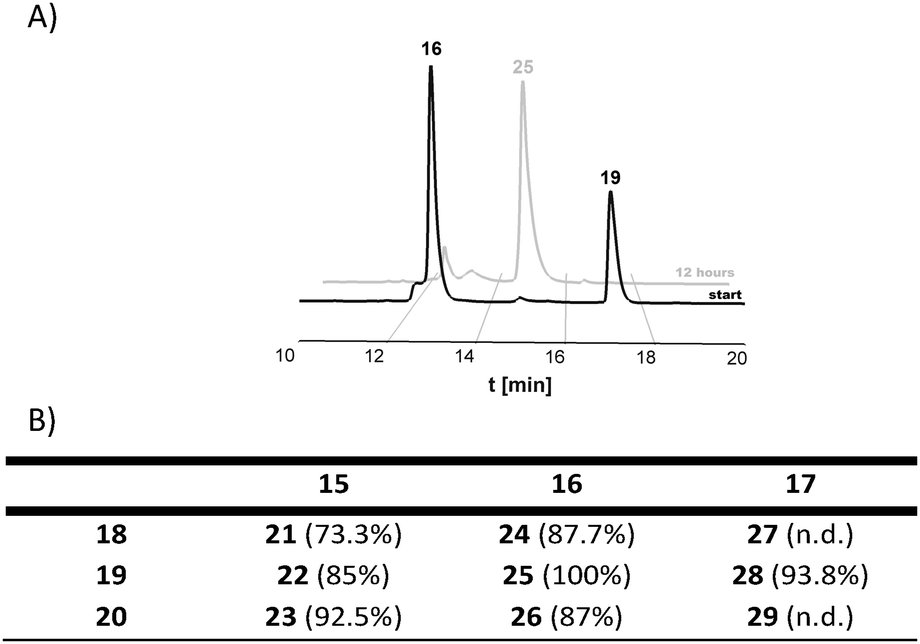 | ||
| Fig. 4 (A) HPLC monitoring of an exemplified DARinv conjugation between Jelleine–Reppe construct 19 and SDMI-3 tetrazine partner 16. (B) Conversion rate for DARinv conjugations calculated from the HPLC traces (n.d.: not determined). | ||
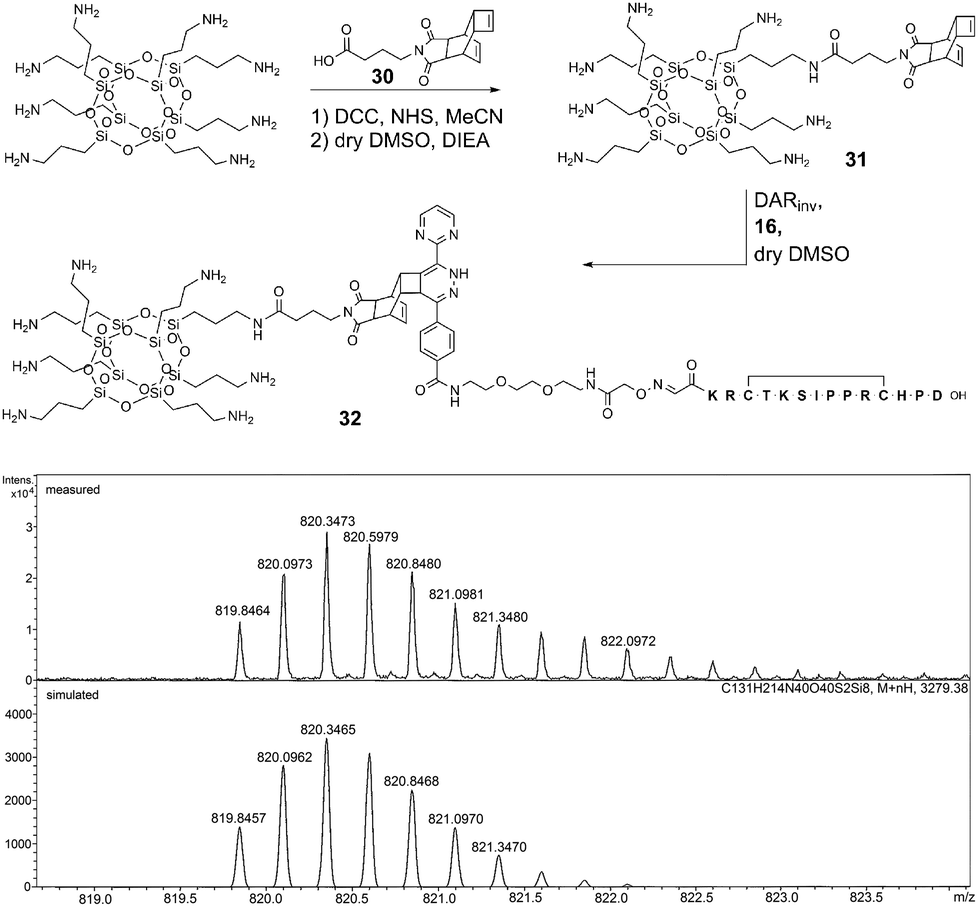 | ||
| Fig. 5 Top: formation of conjugate 32 by DARinv between cell-penetrating COSS 31 and peptide 16. Bottom: exemplary ESI-HR-MS analysis of conjugate 32 [M + 4H]4+. | ||
As the proof-of-concept study showed the viability of our approach, it was further expanded towards a more sophisticated (in view of architecture and functional properties) molecular construct. Thus, we attached a peptidic cargo to the smallest nanoparticle known, a cell-penetrating organic–inorganic hybrid molecule comprising cube-octameric silsesquioxane (COSS).44 This highly symmetric octavalent compound has recently attracted keen attention, being used as a scaffold in a number of biomedical applications, including delivery of bioactive payloads into cancer cells and development of novel tailor-made conjugates of pharmacological interest.44
Compared to the modification of peptides with a dienophile, a different procedure was applied. Thus, Reppe anhydride 2 was equipped with a linker comprising the NHS-activated γ-aminobutyric acid (ESI 1.2.13†). Building block 30 was attached to a single corner of octaamino COSS through an amide bond (Fig. 5 and ESI 1.2.14†) resulting in COSS-dienophile 31 which was reacted with peptide-diene 16 giving conjugate 32. The reaction was carried out in dry DMSO to assure stability of the siloxane core in the presence of the pendant propylamine groups (Fig. 5).
The combination of oxime ligation and DARinv is a convenient method for site-specific conjugation of complex peptidic molecules. What is the rationale behind using the set of two transformations rather than the individual reactions, each of which is bioorthogonal, selective, and fast per se? First, at physiological pH oxime ligation is rather slow and requires catalysis. This could pose a problem if two functional biological macromolecules are ligated directly, whereas the introduction of small bifunctional linker proceeds unhampered, and the next step is irreversible and uncatalyzed. Second, although the direct introduction of DARinv moieties on solid support has been reported for short peptides,21,30,36,51 their introduction in recombinant proteins requires extensive modification of the cellular translation machinery.10,32,52 And, third, our approach provides certain modularity, giving an option to choose between the reactive sites according to reaction environment and nature of the conjugation partners.
In general, our bifunctional building blocks are readily synthetically accessible and can be introduced into peptides site-selectively, without affecting their architecture. The generation of the required carbonyl moieties in peptidic macromolecules is a routine procedure as it proceeds smoothly both upon post-synthetic modification8 and in the course of recombinant production.33 Successive oxime ligation was highly efficient and orthogonal to the side-chain functionalities. Subsequently, the generated DARinv-modified peptides were reacted without a catalyst with the respective counterpart giving defined peptidic constructs with good to quantitative conversion rates. So far, only few DARinv site-specific conjugations of peptides have been reported restricted either to junction of rather short peptidic sequences28 or to the generation of macromolecular constructs of undefined stoichiometry.34,36 Our approach offers the advantage of being suitable for all synthetic peptides, particularly the recombinant ones.
In our hands, being incorporated into peptides and hybrid silsesquioxanes, the strained four-membered ring system of the Reppe anhydride reacted with the respective tetrazine, smoothly converting it into the desired product. This reactivity can be easily explained considering the non-reversible character of DARinv coupling and the fact that cyclobutene is amongst the most reactive species classified via their activation free energies.53
Interestingly, for some peptide conjugates we observed the regeneration of the aromatic system within the tetrazine moiety after DARinv reaction. This observation has previously been reported, but detailed study of the mechanism is still required.49,50
To demonstrate modularity and versatility of our approach, we applied it to covalently graft a bioactive peptide45 onto a cell-penetrating COSS nanoparticle. The success of this reaction opens new avenues for the facile generation of peptide–COSS conjugates for intracellular delivery, thus expanding the toolbox for the chemical modifications of these promising compounds. In a perspective, our system could be applied to the generation of peptide–protein conjugates under physiological conditions.54 Taking into consideration that stability of oxime linkage is context-dependent and at physiological pH a catalyst is required to assure efficient transformation, an adequately high-performance chemistry should be considered to replace this ligation step. To this end, recently reported trapped Knoevenagel condensation55 could be applied as an alternative strategy.
This work was partially funded by the DFG priority program SPP 1623. The authors thank Peter Lorenz (DKFZ, Heidelberg) for his advice regarding tetrazine synthesis.
Notes and references
- A. Angelini and C. Heinis, Curr. Opin. Chem. Biol., 2011, 15, 355–361 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Curr. Org. Chem., 2010, 14, 138–147 CrossRef CAS.
- A. Chakraborty, D. Wang, Y. W. Ebright and R. H. Ebright, Methods Enzymol., 2010, 472, 19–30 CAS.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- B. M. Zeglis, P. Mohindra, G. I. Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder and J. S. Lewis, Bioconjugate Chem., 2011, 22, 2048–2059 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- G. C. Rudolf, W. Heydenreuter and S. A. Sieber, Curr. Opin. Chem. Biol., 2013, 17, 110–117 CrossRef CAS PubMed.
- D. Chelius and T. A. Shaler, Bioconjugate Chem., 2003, 14, 205–211 CrossRef CAS PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- C. H. Kim, J. Y. Axup and P. G. Schultz, Curr. Opin. Chem. Biol., 2013, 17, 412–419 CrossRef CAS PubMed.
- W. P. Heal, S. R. Wickramasinghe, P. W. Bowyer, A. A. Holder, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, Chem. Commun., 2008, 480–482 RSC.
- S. Fabritz, S. Horner, D. Konning, M. Empting, M. Reinwarth, C. Dietz, B. Glotzbach, H. Frauendorf, H. Kolmar and O. Avrutina, Org. Biomol. Chem., 2012, 10, 6287–6293 CAS.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- K. Nwe and M. W. Brechbiel, Cancer Biother. Radiopharm., 2009, 24, 289–302 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G. J. Boons and F. L. van Delft, Angew. Chem., 2010, 49, 3065–3068 CrossRef CAS PubMed.
- N. E. Mbua, J. Guo, M. A. Wolfert, R. Steet and G. J. Boons, ChemBioChem, 2011, 12, 1912–1921 CrossRef CAS PubMed.
- M. Wiessler, W. Waldeck, C. Kliem, R. Pipkorn and K. Braun, Int. J. Med. Sci., 2009, 7, 19–28 Search PubMed.
- R. Pipkorn, W. Waldeck, B. Didinger, M. Koch, G. Mueller, M. Wiessler and K. Braun, J. Pept. Sci., 2009, 15, 235–241 CrossRef CAS PubMed.
- A. Niederwieser, A. K. Spate, L. D. Nguyen, C. Jungst, W. Reutter and V. Wittmann, Angew. Chem., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- S. B. Engelsma, L. I. Willems, C. E. van Paaschen, S. I. van Kasteren, G. A. van der Marel, H. S. Overkleeft and D. V. Filippov, Org. Lett., 2014, 16, 2744–2747 CrossRef CAS PubMed.
- D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H. W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680–13683 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043–8045 RSC.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- S. Ameta, J. Becker and A. Jaschke, Org. Biomol. Chem., 2014, 12, 4701–4707 CAS.
- T. S. Elliott, F. M. Townsley, A. Bianco, R. J. Ernst, A. Sachdeva, S. J. Elsasser, L. Davis, K. Lang, R. Pisa, S. Greiss, K. S. Lilley and J. W. Chin, Nat. Biotechnol., 2014, 32, 465–472 CrossRef CAS PubMed.
- B. M. Zeglis, F. Emmetiere, N. Pillarsetty, R. Weissleder, J. S. Lewis and T. Reiner, ChemistryOpen, 2014, 3, 48–53 CrossRef CAS PubMed.
- J. Seckute and N. K. Devaraj, Curr. Opin. Chem. Biol., 2013, 17, 761–767 CrossRef CAS PubMed.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS PubMed.
- D. Rabuka, J. S. Rush, G. W. deHart, P. Wu and C. R. Bertozzi, Nat. Protoc., 2012, 7, 1052–1067 CrossRef CAS PubMed.
- F. Emmetiere, C. Irwin, N. T. Viola-Villegas, V. Longo, S. M. Cheal, P. Zanzonico, N. Pillarsetty, W. A. Weber, J. S. Lewis and T. Reiner, Bioconjugate Chem., 2013, 24, 1784–1789 CrossRef CAS PubMed.
- K. S. Yang, G. Budin, C. Tassa, O. Kister and R. Weissleder, Angew. Chem., 2013, 52, 10593–10597 CrossRef CAS PubMed.
- R. Hassert, M. Pagel, Z. Ming, T. Haupl, B. Abel, K. Braun, M. Wiessler and A. G. Beck-Sickinger, Bioconjugate Chem., 2012, 23, 2129–2137 CrossRef CAS PubMed.
- J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- R. Rossin, P. R. Verkerk, S. M. van den Bosch, R. C. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., 2010, 49, 3375–3378 CrossRef CAS PubMed.
- J. D. Thomas, H. Cui, P. J. North, T. Hofer, C. Rader and T. R. Burke, Jr., Bioconjugate Chem., 2012, 23, 2007–2013 CrossRef CAS PubMed.
- H. S. Beckmann, A. Niederwieser, M. Wiessler and V. Wittmann, Chemistry, 2012, 18, 6548–6554 CrossRef CAS PubMed.
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- R. G. Kruger, B. Otvos, B. A. Frankel, M. Bentley, P. Dostal and D. G. McCafferty, Biochemistry, 2004, 43, 1541–1551 CrossRef CAS PubMed.
- C. Uth, S. Zielonka, S. Hörner, N. Rasche, A. Plog, H. Orelma, O. Avrutina, K. Zhang and H. Kolmar, Angew. Chem., 2014, 53, 12618–12623 CAS.
- S. Fabritz, S. Horner, O. Avrutina and H. Kolmar, Org. Biomol. Chem., 2013, 11, 2224–2236 CAS.
- H. Fittler, O. Avrutina, M. Empting and H. Kolmar, J. Pept. Sci., 2014, 20, 415–420 CrossRef CAS PubMed.
- B. Glotzbach, M. Reinwarth, N. Weber, S. Fabritz, M. Tomaszowski, H. Fittler, A. Christmann, O. Avrutina and H. Kolmar, PLoS One, 2013, 8, e76956 CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- S. Foillard, M. O. Rasmussen, J. Razkin, D. Boturyn and P. Dumy, J. Org. Chem., 2008, 73, 983–991 CrossRef CAS PubMed.
- A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, J. Org. Chem., 2006, 71, 4903–4909 CrossRef CAS PubMed.
- M. Wiessler, W. Waldeck, R. Pipkorn, C. Kliem, P. Lorenz, H. Fleischhacker, M. Hafner and K. Braun, Int. J. Med. Sci., 2010, 7, 213–223 CrossRef CAS.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- R. Kudirka, R. M. Barfield, J. McFarland, A. E. Albers, G. W. de Hart, P. M. Drake, P. G. Holder, S. Banas, L. C. Jones, A. W. Garofalo and D. Rabuka, Chem. Biol., 2015, 22, 293–298 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Combination of inverse electron-demand Diels–Alder reaction with highly efficient oxime ligation expands the toolbox of site-selective peptide conjugations. See DOI: 10.1039/c5cc03434e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |