
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Real time chemical imaging of a working catalytic membrane reactor during oxidative coupling of methane†
A.
Vamvakeros
ab,
S. D. M.
Jacques
*bc,
V.
Middelkoop
*d,
M.
Di Michiel
e,
C. K.
Egan
c,
I. Z.
Ismagilov
f,
G. B. M.
Vaughan
e,
F.
Gallucci
g,
M.
van Sint Annaland
g,
P. R.
Shearing
h,
R. J.
Cernik
c and
A. M.
Beale
*ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: andrew.beale@ucl.ac.uk
bUK Catalysis Hub, Research Complex at Harwell, Harwell, Didcot, Oxon OX11 0FA, UK
cSchool of Materials, University of Manchester, Manchester, Lancashire M13 9PL, UK. E-mail: simon.jacques@gmail.com
dFlemish Institute for Technological Research, VITO NV, Boeretang 200, Mol, Belgium. E-mail: vesna.middelkoop@vito.be
eESRF, 6 Rue Jules Horowitz, 38000 Grenoble, France
fBoreskov Institute of Catalysis SB RAS, Pr. Akademika Lavrentieva 5, 630090 Novosibirsk, Russia
gChemical Process Intensification, Department of Chemical Engineering, Eindhoven University of Technology, Box 315, Eindhoven, Netherlands
hDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
First published on 28th May 2015
Abstract
We report the results from an operando XRD-CT study of a working catalytic membrane reactor for the oxidative coupling of methane. These results reveal the importance of the evolving solid state chemistry during catalytic reaction, particularly the chemical interaction between the catalyst and the oxygen transport membrane.
Ethylene is an important platform chemical and with the closure of naphtha crackers, new processes for its production are required. The continuous reduction of gas flaring and the exploitation of shale gas by hydraulic fracturing (fracking) has led to a dramatic increase in the availability of methane.1 Natural gas, whose main component is methane, is considered to be an abundant hydrocarbon source compared to crude oil and there is much interest in producing higher value bulk chemicals from it.2 Environmentally-friendly and cost-effective processing technologies for direct conversion of methane to light olefins (e.g. ethylene) as an alternative to the highly energy-intensive cracking of crude oil are needed.2b,3 The oxidative coupling of methane (OCM) can potentially provide an economically viable route for ethylene production.4 Additionally, the application of catalytic membrane reactors (CMR) employing oxygen transport membranes can lead to a decrease of the cost of the overall process.5 Importantly, recent research has shown that CMRs for the OCM can achieve ∼35% C2 yield; one of the highest values reported in literature.6
In this study, we demonstrate the application of high energy X-ray diffraction computed tomography (XRD-CT) to investigate the chemical evolution of a working CMR for the OCM reaction. This is the first solid state study under operando conditions and uses state-of-the-art OCM catalyst (Mn–Na–W/SiO2) and oxygen transport membrane (BaCoxFeyZrzO3−δ). XRD-CT allowed us to observe the catalyst and membrane in their active states and revealed the presence of a new phase (i.e. BaWO4) forming at the interface between the catalyst particles and the membrane under high temperatures which may impact on the long-term stability and performance of this integrated reactor system. We are able to obtain this unprecedented chemical information because XRD-CT enables local signals to be obtained from within bulk dense objects; conventional methods such as powder XRD are “blind” to such local heterogeneities.7 XRD-CT is one of a series of recently developed X-ray-based techniques which provide spatially-resolved ‘chemical’ insight and can be employed under process conditions to obtain a more comprehensive appreciation of structure–activity relationships in catalytic/functional materials.8
The OCM and the direct non-oxidative conversion of methane are the two most promising methane-to-olefins conversion technologies as they require a single catalytic reactor compared to conventional gas-to-liquid (GTL) chemical plants where two reactors are used in series (i.e. the first for steam reforming of methane and the second for Fischer–Tropsch synthesis).9 Currently there are long-term stability problems with catalysts used in non-oxidative conversion of methane in contrast to the Mn–Na–W/SiO2 OCM catalyst that has shown high stability for hundreds of hours of operation.10 This catalyst is considered to be one of the most promising for the OCM reaction mainly due to its long term stability and productivity under high temperatures (i.e. more than 80% selectivity for C2 molecules for 20% methane conversion).11 Despite more than twenty years extensive study, there is still much debate as to the nature of the phases/species present in the Mn–Na2WO4/SiO2 catalyst under reaction conditions. For example, in a recent review paper by Arndt et al., it was concluded that some form of synergy must occur between the various Mn, Na and W containing phases/species present which must be responsible for the catalyst activity.12
The main drawbacks of the OCM reaction are the total oxidation of methane and/or the conversion of formed C2 molecules to COx which is difficult to avoid in traditional fixed bed reactors at high conversions.13 One way to potentially overcome this problem is to use CMRs which regulate the oxygen flow to the catalyst.6,14 The membranes used in these CMRs are mixed ionic and electronic conducting (MIEC) materials that allow only oxygen ions to diffuse through them.15 Especially in the case of the OCM, it seems that CMRs present the only solution to the intrinsic problems of this reaction.16 By maintaining a low oxygen partial pressure on the methane ‘side’ and by homogeneously distributing the oxygen ions over the reactor, improved C2 selectivity is attained and the further oxidation of the C2 molecules can be mitigated. Several perovskite-type (formula of ABO3−δ) MIEC membranes have been tested as catalysts for the OCM reaction per se in disk, tubular and hollow-fiber geometries.15b,16c,17 However, it has been shown that the combination of a MIEC membrane with a catalyst bed leads to higher C2 yield at lower temperatures.16a,b BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ) hollow fiber membranes have proven to be a good compromise between the required chemical stability (i.e. stability of the perovskite structure under different environments) and oxygen permeation flux.16b,18 Therefore, these materials (i.e. the BCFZ membrane and the Mn–Na2WO4/SiO2 catalyst) are promising but need to be studied further in order to determine their prospects as the most appropriate components for the CMR.
Two XRD-CT experiments were performed using the same CMR (i.e. BCFZ with Mn–Na–W/SiO2 catalyst) at station ID15A of the ESRF (Fig. S1, ESI†). In these XRD-CT experiments, air was used at the outer side of the membrane in all cases (i.e. flow rate of 100 mL min−1). In the first experiment, an in situ study, which will be referred to as OCM1, the temperature of the system was increased under the flow of methane (5 vol% CH4 in He at 100 mL min−1). In the second experiment, OCM2, the temperature of the system was increased under the flow of He (flow rate of 30 mL min−1). Air was then used to ensure the optimal state of the catalyst in terms of activity (flow rate of 50 mL min−1). Finally, different mixtures of CH4 diluted in He were used for the actual OCM experiment (5, 20, 50 and 100 vol% CH4 in He while the total flow rate was kept constant at 100 mL min−1).
A schematic representation of the CMR is shown at the top left of Fig. 1; this illustration is produced from absorption computed tomography (micro-CT) data collected before the OCM experiment. XRD-CT was performed at one fixed position, vertically at the middle of the reactor. At the bottom right of Fig. 1, reconstructed XRD-CT images of raw scattered intensity at scattering angle 1.75° are presented. These show the growth of the SiO2 tridymite phase during the operation of the CMR but serve to illustrate that evolving solid-state chemistry can be followed. Each XRD-CT reconstructed slice is actually a data cube where each composing voxel (a three dimensional pixel) contains a reconstructed diffraction pattern (see also comment 1 in the ESI†).
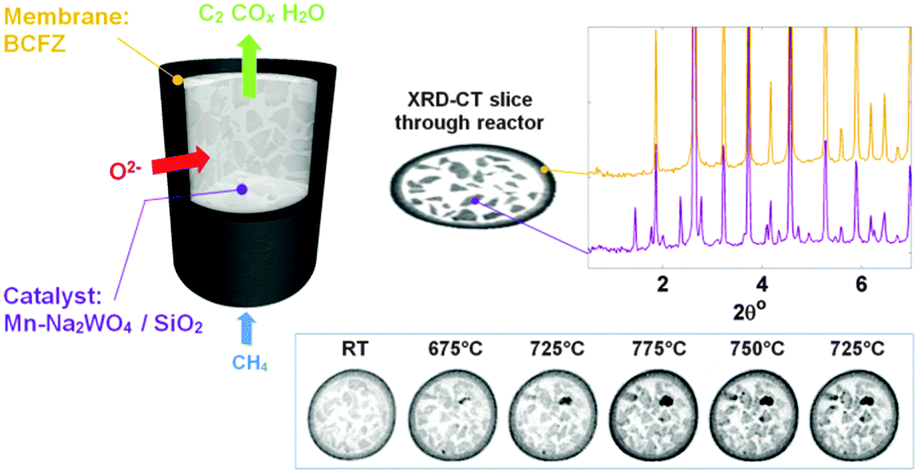 | ||
| Fig. 1 Left: image showing the CMR. Top right: within an XRD-CT image each pixel has a corresponding diffraction pattern; the graph inset top right shows two selected diffraction patterns: one from the membrane (yellow trace) and one from the catalyst region (purple trace). Images can be constructed that correspond to features (i.e. Bragg reflections) within the diffraction patterns. Bottom right: maps of raw scattered intensity at scattering angle 1.75° 2θ corresponding to the tridymite phase. Images are displayed with a common intensity scaling. | ||
Fig. 2 shows the evolution of the catalyst as the OCM1 experiment proceeds, presented as phase maps showing the distributions of five identified crystalline phases and one amorphous phase at all temperatures where XRD-CT data were measured. Also shown is the membrane (derived from a map of the intensity of the (100) BCFZ reflection) though its distribution does not change during the course of the experiment; we do however observe some relative changes in the reflection intensities for BCFZ as described in the ESI† (Fig. S2). At room temperature, crystalline Mn2O3 and Na2WO4 are present in the catalyst particles as expected. The SiO2 support is clearly visible, principally in α-cristobalite and secondarily in tridymite forms. More importantly, the distributions of Mn2O3 and Na2WO4 show that they are not perfectly co-located. At 675 °C, the Na2WO4 phase disappears, which is in agreement with the work of Hou et al., and is coincident with the appearance of an amorphous phase.19 However, a new phase is seen to appear which we identify as BaWO4 (using the ICSD and the ICCD PDF crystallographic databases (Fig. S3, ESI†)). The distribution of BaWO4 does not match the starting distributions of Na2WO4 but instead concentrated spots of this phase form predominantly at the catalyst/membrane interface. At 725 °C, Mn2O3 decreases but significant amounts of BaWO4 grow which appears to migrate towards the inner wall of the membrane. On reaching 775 °C, Mn2O3 is barely detectable yet more BaWO4 forms, lining almost the entirety of the inner membrane wall. We note, Mn–BaWO4/SiO2 has been tested in the past as an OCM catalyst and shows higher selectivity to COx than to C2 molecules which suggests that the formation of this phase is likely to be detrimental to catalytic performance.20 On reducing the temperature to 725 °C, Mn2O3 is seen to reform. Against the backdrop of these changes, the SiO2 support is seen to also evolve with continuous growth of tridymite (more than doubling in quantity) and less significant initial growth then loss/conversion of cristobalite from tetragonal to cubic forms; this is also in agreement with the work of Hou et al. (i.e. α-β transition).19
 | ||
| Fig. 2 Phase maps for BCFZ, cristobalite and tridymite (SiO2 polymorphs), Na2WO4, Mn2O3, Ba2WO4 and an amorphous phase as determined from the XRD-CT data. These maps have been obtained from the integrated intensities of the respective phases. The relative change of each component, as determined from the integrated intensities (for the whole sample slice), is shown in Fig. S4 (ESI†). | ||
In Fig. 3, the results from the OCM2 experiment are presented. It is essential to note that in both OCM experiments (i.e. OCM1 and OCM2) the same solid-state chemistry takes place demonstrating that the results from the OCM1 experiment are reproducible. In panel G of Fig. 3, it is shown that when 5 vol% CH4 diluted in He (total flow rate of 100 mL min−1) is used, there is a new signal in the mass spectrometry data (m/z 26) which corresponds to C2 fractions. The profiles of selected masses throughout the experiment are presented in Fig. S5 (ESI†) where it is shown that C2 fractions increase with increasing CH4 concentration, and that C3 fractions are also forming. It is therefore reasonable to assume that the catalyst was captured in its active state in both experiments. The mass spectrometry data also suggest that there is no short term loss in activity with formation of BaWO4. The observation that there is no simultaneous loss of activity when W is being locked away (as BaWO4) suggests there is Na2WO4 present in the system which is not participating as part of the active catalyst.
 | ||
| Fig. 3 Panels A and B show reconstructed slices from micro-CT measurements of the CMR before and after the OCM2 experiment at room temperature. Panels C and D are the same images repeated with the distribution of cristobalite as found from XRD-CT overlaid in violet. Panels E and F again contain the micro-CT slices but here overlaid with the distributions of Na2WO4 (red) and BaWO4 (green). Panel G shows the mass spectrometry data that correspond to the following masses (m/z): 4 (He – cyan), 15 (CH4 – blue), 28 (N2, CO, C2H4+ – green), 32 (O2 – red), 44 (CO2 – black) and 26 (C2H2+ – magenta). Panel H shows the overlaid WDS elemental maps for Ba (grayscale) and W (green). Panel I shows a summed diffraction pattern from selected pixels at the membrane/catalyst interface. Overlaid on this pattern are sticks of database intensities for phases BaWO4 (green), cristobalite (magenta), Mn2O3 (blue), tridymite (yellow) and BCFZ (grey). | ||
From the OCM2 experiment we observe that Na2WO4 reforms during cooling after the OCM experiment, albeit in lower concentrations to that of the initial state, and that the formed BaWO4 persists, as does the distribution and final forms of the support material (panels E and F in Fig. 3). This is also apparent in the micro-CT data where the hotspots (the overlaid red and green circles highlight example hotspot regions in panels A and B of Fig. 3) are large concentrations of the W-containing phases (Fig. S6, ESI†).
Our observations regarding the BaWO4 phase are confirmed by ex situ SEM/WDS elemental mapping (Fig. S7, ESI†). In panel H of Fig. 3, the elemental maps of Ba and W at a cross section of the CMR are presented after overlaying the two images. W is seen to be co-located with Ba at the edge the inner side of the membrane wall. Panel I shows a summed diffraction pattern from selected pixels at the membrane/catalyst interface where the following phases have been identified: BaWO4, cristobalite, Mn2O3, tridymite and BCFZ.
In this study we have shown that spatially resolved diffraction information can be gleaned from within the confines of a working reactor and that the results are reproducible. Indeed the state of the catalyst was captured prior to, under and after OCM operando conditions. We have confirmed the initial catalyst contains both Mn2O3 and Na2WO4 and shown these phases are not perfectly co-located. Na2WO4 is seen to melt (consistent with previous observations) and coincident to this event we observed the formation of an amorphous component.19 The diffraction signal from Mn2O3 is seen to decrease approaching the higher temperatures required for OCM. A new stable phase, BaWO4 is seen to form at the interface between the catalyst particles and the inner side of the BCFZ membrane (the mobility of W species was independently confirmed by micro-CT and ex situ elemental mapping). We also observed significant evolution of the silica support phases which will impact on available surface area of the catalyst. Under OCM conditions, significant quantities of cristobalite and tridymite are present as is an amorphous phase and BaWO4. There are not significant quantities of highly crystalline Mn2O3 under OCM but the loss of highly crystalline Mn2O3 was not accompanied by any readily observable new amorphous component. Mn2O3 and Na2WO4 reform (albeit in lower final concentrations) post OCM whereas the formed BaWO4 persists. The observation of the formation of a stable BaWO4 phase is important since it demonstrates that the high and uncontrolled mobility of W at high temperatures is problematic in that: (1) the loss of W could impact on the long-term stability of the catalyst, (2) it will almost certainly lead to the deactivation of the membrane after many hours of operation by forming a layer at the membrane wall and therefore blocking the flow of oxygen to the catalyst bed, (3) and its presence will likely affect the reactor performance as Mn–BaWO4/SiO2 shows high selectivity to COx.20 Beyond our experimental findings for the CMR and its use for the OCM reaction, this also work illustrates the value of these types of measurements in evaluating the performance of a reactor system for a chemical reactor engineering-based problem.
The development of the catalysts and membranes for the catalytic membrane reactor used in this work is funded within the DEMCAMER project as part of the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. NMP3-LA-2011-262840. Note: “The present publications reflect only the authors' views and the Union is not liable for any use that may be made of the information contained therein.” The authors would like to thank the European Synchrotron Radiation Source for beam time and CerPoTech AS for providing the ceramic powders. The authors would like to acknowledge support from the Hercules Foundation (project ZW09-09) for providing access to the FEG-EPMA at MTM-Catholic University of Leuven. Simon Jacques is supported under the EPSRC RCaH Impact Acceleration Fellowship. The authors Andrew M. Beale and Antonios Vamvakeros are also supported by the EPSRC funding. Paul R. Shearing acknowledges funding from the Royal Academy of Engineering.
Notes and references
- R. A. Kerr, Science, 2010, 328, 1624 CrossRef CAS PubMed.
- (a) J. H. Lunsford, Catal. Today, 2000, 63, 165 CrossRef CAS; (b) E. McFarland, Science, 2012, 338, 340 CrossRef CAS PubMed.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580 CAS.
- H. R. Godini, S. Xiao, S. Jašo, S. Stünkel, D. Salerno, N. X. Son, S. Song and G. Wozny, Fuel Process. Technol., 2013, 106, 684 CrossRef CAS PubMed; Y. Khojasteh Salkuyeh and T. A. Adams, II, Energy Convers. Manage., 2015, 92, 406 CrossRef PubMed.
- J. A. Hugill, F. W. A. Tillemans, J. W. Dijkstra and S. Spoelstra, Appl. Therm. Eng., 2005, 25, 1259 CrossRef PubMed.
- S. Bhatia, C. Y. Thien and A. R. Mohamed, Chem. Eng. J., 2009, 148, 525 CrossRef CAS PubMed.
- G. Harding, J. Kosanetzky and U. Neitzel, Med. Phys., 1987, 14, 515 CrossRef CAS; P. Bleuet, E. Welcomme, E. Dooryhée, J. Susini, J. L. Hodeau and P. Walter, Nat. Mater., 2008, 7, 468 CrossRef PubMed; S. D. M. Jacques, M. Di Michiel, A. M. Beale, T. Sochi, M. G. O'Brien, L. Espinosa-Alonso, B. M. Weckhuysen and P. Barnes, Angew. Chem., Int. Ed., 2011, 50, 10148 CrossRef PubMed; M. G. O'Brien, S. D. M. Jacques, M. Di Michiel, P. Barnes, B. M. Weckhuysen and A. M. Beale, Chem. Sci., 2012, 3, 509 RSC; A. M. Beale, E. K. Gibson, M. G. O'Brien, S. D. M. Jacques, R. J. Cernik, M. D. Michiel, P. D. Cobden, Ö. Pirgon-Galin, L. V. D. Water, M. J. Watson and B. M. Weckhuysen, J. Catal., 2014, 314, 94 CrossRef PubMed; A. M. Beale, S. D. M. Jacques, E. K. Gibson and M. Di Michiel, Coord. Chem. Rev., 2014, 277, 208 CrossRef PubMed.
- F. Basile, P. Benito, S. Bugani, W. De Nolf, G. Fornasari, K. Janssens, L. Morselli, E. Scavetta, D. Tonelli and A. Vaccari, Adv. Funct. Mater., 2010, 20, 4117 CrossRef CAS PubMed; I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873 CrossRef PubMed; J.-D. Grunwaldt, J. B. Wagner and R. E. Dunin-Borkowski, ChemCatChem, 2013, 5, 62 CrossRef PubMed; S. D. M. Jacques, M. Di Michiel, S. A. Kimber, X. Yang, R. J. Cernik, A. M. Beale and S. J. Billinge, Nat. Commun., 2013, 4, 2536 Search PubMed; S. W. Price, K. Ignatyev, K. Geraki, M. Basham, J. Filik, N. T. Vo, P. T. Witte, A. M. Beale and J. F. Mosselmans, Phys. Chem. Chem. Phys., 2015, 17, 521–529 RSC.
- J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1995, 34, 970 CrossRef CAS PubMed; Q. Zhu, S. L. Wegener, C. Xie, O. Uche, M. Neurock and T. J. Marks, Nat. Chem., 2013, 5, 104 CrossRef PubMed; X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616 CrossRef PubMed; M. Ruitenbeek and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 11137 CrossRef PubMed; P. Schwach, M. G. Willinger, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11381 CrossRef PubMed.
- X. Fang, S. Li, J. Lin and Y. Chu, J. Mol. Catal., 1992, 6, 427 Search PubMed; X. P. Fang, S. B. Li, J. Z. Lin, J. F. Gu and D. X. Yang, J. Mol. Catal., 1992, 6, 255 Search PubMed; Y. Kou, B. Zhang, J.-z. Niu, S.-b. Li, H.-l. Wang, T. Tanaka and S. Yoshida, J. Catal., 1998, 173, 399 CrossRef CAS; J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792 RSC.
- D. J. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1995, 155, 390 CrossRef CAS.
- S. Arndt, T. Otremba, U. Simon, M. Yildiz, H. Schubert and R. Schomäcker, Appl. Catal., A, 2012, 425–426, 53 CrossRef CAS PubMed.
- L. Luo, X. Tang, W. Wang, Y. Wang, S. Sun, F. Qi and W. Huang, Sci. Rep., 2013, 3, 1625 Search PubMed; K. Takanabe and E. Iglesia, Angew. Chem., Int. Ed., 2008, 47, 7689 CrossRef CAS PubMed.
- H. Wang, Y. Cong and W. Yang, Catal. Today, 2005, 104, 160 CrossRef CAS PubMed.
- (a) Y. Teraoka, H.-M. Zhang, S. Furukawa and N. Yamazoe, Chem. Lett., 1985, 1743 CrossRef CAS; (b) X. Dong, W. Jin, N. Xu and K. Li, Chem. Commun., 2011, 47, 10886 RSC.
- (a) X. Tan, Z. Pang, Z. Gu and S. Liu, J. Membr. Sci., 2007, 302, 109 CrossRef CAS PubMed; (b) O. Czuprat, T. Schiestel, H. Voss and J. Caro, Ind. Eng. Chem. Res., 2010, 49, 10230 CrossRef CAS; (c) X. Tan and K. Li, Curr. Opin. Chem. Eng., 2011, 1, 69 CrossRef CAS PubMed.
- Z. Cao, H. Jiang, H. Luo, S. Baumann, W. A. Meulenberg, J. Assmann, L. Mleczko, Y. Liu and J. Caro, Angew. Chem., Int. Ed. Engl., 2013, 52, 13794 CrossRef CAS PubMed; Y. Wei, W. Yang, J. Caro and H. Wang, Chem. Eng. J., 2013, 220, 185 CrossRef PubMed; H. Jiang, Z. Cao, S. Schirrmeister, T. Schiestel and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 5656 CrossRef PubMed; J. E. ten Elshof, B. A. van Hassel and H. J. M. Bouwmeester, Catal. Today, 1995, 25, 397 CrossRef; J. E. ten Elshof, H. J. M. Bouwmeester and H. Verweij, Appl. Catal., A, 1995, 130, 195 CrossRef; S. J. Xu and W. J. Thomson, AIChE J., 1997, 43, 2731 CrossRef PubMed; Y. Zeng, Y. S. Lin and S. L. Swartz, J. Membr. Sci., 1998, 150, 87 CrossRef; W. Wang and Y. S. Lin, J. Membr. Sci., 1995, 103, 219 CrossRef; X. Tan and K. Li, Ind. Eng. Chem. Res., 2006, 45, 142 CrossRef; F. R. García-García, B. F. K. Kingsbury, M. A. Rahman and K. Li, Catal. Today, 2012, 193, 20 CrossRef PubMed.
- J. Tong, W. Yang, B. Zhu and R. Cai, J. Membr. Sci., 2002, 203, 175 CrossRef CAS; T. Schiestel, M. Kilgus, S. Peter, K. J. Caspary, H. Wang and J. Caro, J. Membr. Sci., 2005, 258, 1 CrossRef PubMed; C. Tablet, G. Grubert, H. Wang, T. Schiestel, M. Schroeder, B. Langanke and J. Caro, Catal. Today, 2005, 104, 126 CrossRef PubMed; K. Efimov, O. Czuprat and A. Feldhoff, J. Solid State Chem., 2011, 184, 1085 CrossRef PubMed; S. M. Hashim, A. R. Mohamed and S. Bhatia, Adv. Colloid Interface Sci., 2010, 160, 88 CrossRef PubMed.
- S. Hou, Y. Cao, W. Xiong, H. Liu and Y. Kou, Ind. Eng. Chem. Res., 2006, 45, 7077 CrossRef CAS.
- S. Ji, T. Xiao, S. Li, L. Chou, B. Zhang, C. Xu, R. Hou, A. P. E. York and M. L. H. Green, J. Catal., 2003, 220, 47 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc03208c |
| This journal is © The Royal Society of Chemistry 2015 |