
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guanosine-based hydrogen-bonded 2D scaffolds: metal-free formation of G-quartet and G-ribbon architectures at the solid/liquid interface†
Mohamed El
Garah‡
a,
Rosaria C.
Perone‡
b,
Alejandro Santana
Bonilla
cd,
Sébastien
Haar
a,
Marilena
Campitiello
b,
Rafael
Gutierrez
c,
Gianaurelio
Cuniberti
*ce,
Stefano
Masiero
*b,
Artur
Ciesielski
*a and
Paolo
Samorì
*a
aISIS & icFRC, Université de Strasbourg & CNRS, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: samori@unistra.fr; ciesielski@unistra.fr
bAlma Mater Studiorum – Università di Bologna, Dipartimento di Chimica “G. Ciamician”, Via S. Giacomo 11, 40126 Bologna, Italy. E-mail: stefano.masiero@unibo.it
cInstitute for Materials Science and Max Bergmann Center of Biomaterials, Dresden University of Technology, 01062 Dresden, Germany
dMax Planck Institute for the Physics of Complex Systems, 01187 Dresden, Germany
eCenter for Advancing Electronics Dresden, Dresden Center for Computational Materials Science, Dresden University of Technology, 01062 Dresden, Germany. E-mail: g.cuniberti@tu-dresden.de
First published on 9th June 2015
Abstract
We report on the synthesis and self-assembly of three novel lipophilic guanosine derivatives exposing a ferrocene moiety in the C(5′) position of the sugar unit. Their self-association in solution, and at the solid/liquid interface, can be tuned by varying the size and nature of the C(8)-substituent, leading to the generation of either G-ribbons, lamellar G-dimer based arrays or the G4 cation-free architectures.
The controlled self-assembly of suitably designed molecular building blocks is a viable approach towards the construction of highly sophisticated nanostructured materials.1 Among various molecular components, supramolecular architectures with ad hoc structural motifs can be obtained through the non-covalent self-association of natural2 and unnatural3 nucleobases on flat surfaces. Such structures, when decorated with appropriate electrically/optically active units, can be used as scaffolds to locate such units in pre-determined positions in 2D,4 thereby paving the way towards a wide range of applications, e.g. in opto-electronics.5 Among the four nucleobases of DNA, guanine (G)6 exhibits a very rich self-assembly behaviour: depending on the environmental conditions it can undergo different self-assembly pathways resulting in various well-distinct architectures including dimers,7 tetramers,8 ribbons,9 and helical structures.10 In the presence of certain metal ions, G can form cyclic tetrameric architectures, also known as G-quartets (hereafter G4), which further pile up into octamers or higher order columnar aggregates. It is commonly believed that templating alkali metal cations such as Na+ and K+ as well as alkaline earth and lanthanide cations are needed to stabilize the G4 formation.10 However, suitably designed guanosines, e.g. derivatives exposing a sterically demanding N,N-dimethylaniline moiety in the C(8) position of the guanine core, were found to form cation-free G4 structures both in solution and in the solid state of the bulk.11
On solid surfaces, G-based H-bonded supramolecular architectures were self-assembled into highly ordered motifs and studied by scanning tunnelling microscopy (STM) under ultra-high vacuum.12 However, the STM explorations at the solid/liquid interfaces have shown numerous advantages, e.g. they provide an excellent environment for in situ chemical modifications of adsorbed molecules.13 When guanosine derivatives are physisorbed at the solution/graphite interface, thermodynamically stable supramolecular ribbons, characterized by N(2)–H⋯O(6) and N(1)–H⋯N(7) H-bonds, were observed.
Given the possibility of functionalizing the guanosines in the sugar moiety, they represent ideal building blocks for the fabrication of conformationally rigid and structurally complex architectures based on ribbons or G4 motifs. Yet, the formation of G4 at the solid/liquid interface was observed only upon using a templating metal center.2c
Ferrocenes are organometallic compounds possessing unique opto-electronic properties, which made them important active components for applications in medicine and materials science. In this context, the control over the self-assembly of ferrocene-based architectures through molecular engineering is crucial in order to control and improve their opto-electronic properties.
Here we have designed and synthesized three novel lipophilic guanosine derivatives G1–G3 (see Scheme 1 and the ESI† for synthetic details), exposing a ferrocene moiety in the C(5′) position of the sugar unit.
 | ||
| Scheme 1 Chemical formulae of the investigated guanosine derivatives. | ||
In order to tune the molecular self-assembly process at the graphite−solution interface we substituted the nucleobase C(8) position with different sterically demanding groups. The presence of a long stearate side chain in the C(3′) position of the sugar unit is expected to promote the molecular physisorption on HOPG.
In line with previous studies on other guanosines,2a,b in the absence of metal ions, G1 in solution forms an H-bonded ribbon-like structure that involves the pairing N(2)–H⋯O(6) and N(1)–H⋯N(7). 1H NMR spectra in CDCl3 (Fig. S1 in the ESI†) show a progressive downfield shift of both N(1)–H and N(2)–H signals upon cooling, while considerable line broadening occurs (e.g. see the C(8)–H signal at δ = 7.8 in Fig. S1, ESI†). G1 can complex alkali metal ions to form a C4 symmetric octamer consisting of two stacked G14, as evidenced from the characteristic changes both in the 1H NMR and in the CD spectrum (Fig. S2, ESI†). The self-assembly of G1 at the solid–liquid interface has been explored by applying a 4 μL drop of a (100 ± 2) μM G1 solution in 1-phenyloctane on HOPG. The STM image showed a crystalline structure consisting of ribbon-like architectures forming a lamellar motif (Fig. 1a). In this 2D crystal, the stearate side chains are physisorbed flat on the surface and they are interdigitated between adjacent lamellae. The unit cell parameters amount to a = (7.4 ± 0.1) nm, b = (1.0 ± 0.1) nm, and α = (88 ± 2)°, leading to an area A = (7.4 ± 0.2) nm2, where each unit cell contains four molecules. Thus, the area occupied by a single molecule G1 corresponds to (1.85 ± 0.10) nm2. Given the size of the unit cell there is not enough space to accommodate the ferrocene units on the basal plane of the HOPG surface, thus it is most likely that they are either back-folded into supernatant solution or physisorbed as a second layer on top of the guanosine first layer. Unfortunately, despite the high spatial resolution achieved by STM imaging, we are unable to rule out any of these two scenarios. The monitored supramolecular motif can be well-described by the formation of a 1D hydrogen-bonded ribbon that involves the pairing N(2)–H⋯O(6) and N(1)–H⋯N(7) (see the model in Fig. 1b and Fig. S12, ESI†). This self-assembly behaviour is in good agreement with NMR solution data.
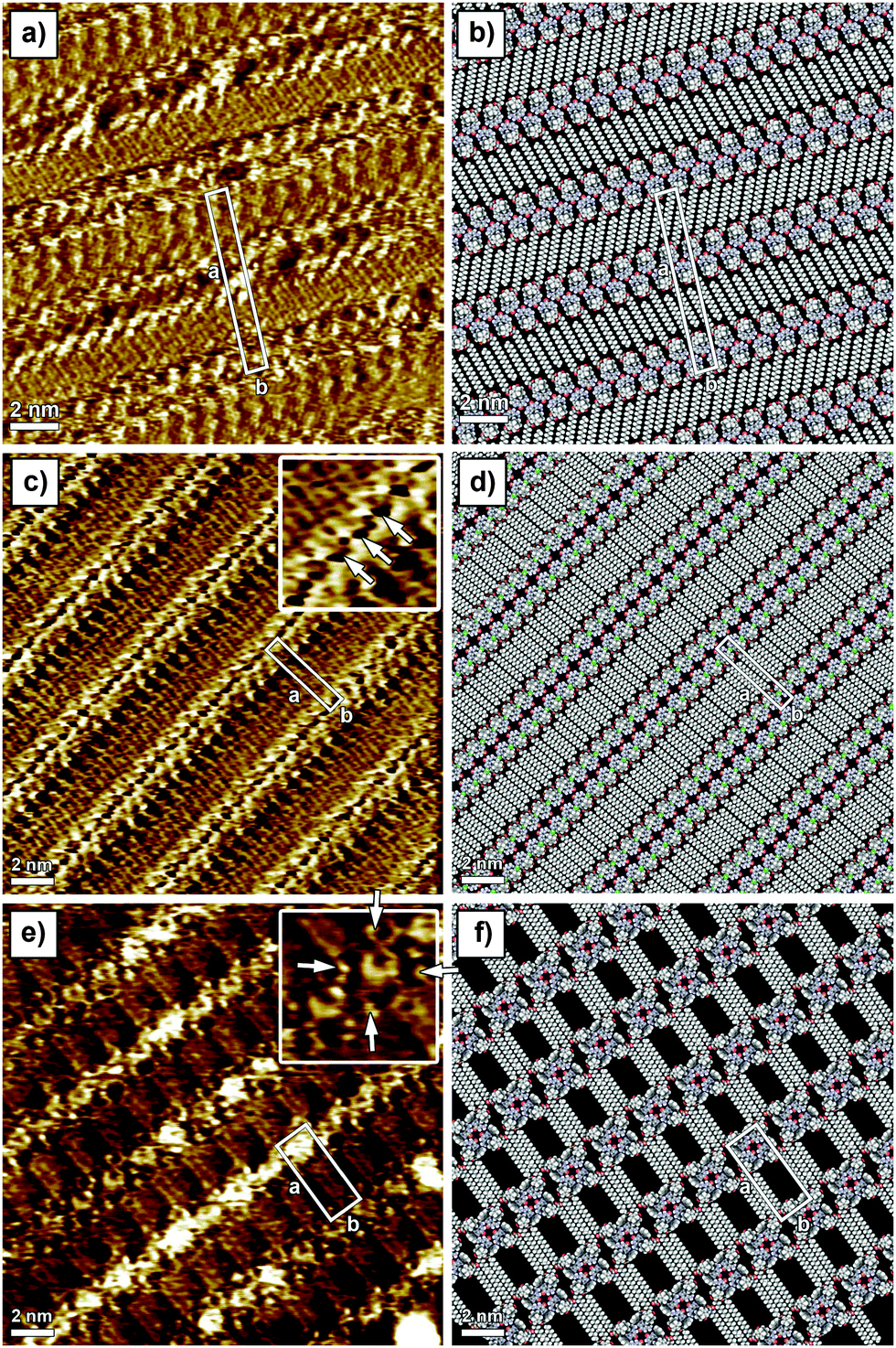 | ||
| Fig. 1 STM images of physisorbed monolayers of the investigated guanosine derivatives self-assembled on HOPG from solutions in 1-phenyloctane. Ribbon-like structure of (a) G1 and (c) G2. (e) G4-based structure of G3. The packing motifs are shown in (b), (d) and (f), respectively. Tunneling parameters in (a), (c) and (e): average tunnelling current (It) = (35 ± 2) pA, bias voltage (Vt) = (400 ± 25) mV. | ||
In order to steer the G self-assembly towards different supramolecular motifs, we explored the effect of the functionalization of the C(8) position of the guanine core, by substituting the proton with a Br atom (G2). Monolayers of G2 have been generated by applying on the HOPG surface a 4 μL drop of a (100 ± 2) μM solution of G2 in 1-phenyloctane. The STM imaging (Fig. 1c) displays a crystalline structure consisting of lamellar architectures. In a G2-based 2D crystal, the stearate side chains are physisorbed flat on the surface and are interdigitated between adjacent lamellae. The unit cell parameters, a = (4.1 ± 0.1) nm, b = (0.9 ± 0.1) nm, and α = (90 ± 2)°, lead to an area A = (3.7 ± 0.1) nm2, where each unit cell contains two molecules. Thus, the area occupied by a single molecule G2 amounts to (1.85 ± 0.10) nm2. While the area occupied by single molecule G2 is identical to that of G1, their self-assembled patterns are markedly different (see Fig. 1a vs. c). In particular, the appearance of hollow features within the G2 ribbon core (indicated with arrows in the inset of Fig. 1c) as well as different orientations of stearate side chains vs. the main lamellar axis (60° and 90° for G1 and G2 patterns, respectively) provides unambiguous evidence for a different self-assembly motif. In fact, the G2 supramolecular motif can be well-described by the formation of H-bonded dimers, which involves the pairing N(1)–H⋯O(6) (see models in Fig. 1d and 2). Each dimer interacts laterally with neighbouring dimers via N(2)–H⋯Br(8) bonding, resulting in the formation of 1D lamellar arrays. Similarly to the case of G1 ribbons, ferrocene units are likely back-folded into supernatant solution or adsorbed as a second layer. Formation of such structures highlights the role played by bulky bromine atoms in the C(8) position of the G core, which introduced N(2)–H⋯Br(8) bonding, thus forcing self-assembly towards an unprecedented ribbon structure.
 | ||
| Fig. 2 Calculated structures of G1 H-bonded ribbon, G2 lamella and G34 cation-free quartet. | ||
1H-NMR spectra of G2 recorded upon cooling a solution in CDCl3 (Fig. S3, ESI†) show a progressive splitting of the broad N(2)–H singlet at δ = 6.1 into two signals (bonded and free N(2)–H, at δ = 8.7 and δ = 5.7, respectively). The chemical shifts for the N(2)–H protons are close to those reported for a similar compound (δ = 8.50 and δ = 5.44),10b but differ from those of the two stacked G4 formed by G1 in the presence of metal ions (marked signals in Fig. S1, ESI†) as well as from those of an isolated G4 (δ = 9.81 and δ = 5.15).11 Previously,10b some of us have studied the self-assembly of a similar 8-bromo lipophilic guanosine derivative in solution by NMR spectroscopy. It was concluded that this signal splitting can be attributed to the existence of isolated G4 on the basis of the well-known preference of 8-bromo guanosine to adopt a syn conformation around the glycosidic bond and the lack of any liquid crystalline behavior. Although G2 behaves similarly and the lack of substantial line broadening points to the existence of small size aggregates, the present results suggest to reconsider the supramolecular behavior of G2 in solution, taking into account the existence of small, possibly dimer-like, aggregates.
We then decided to replace the Br atom with a more neutral group, which is also more sterically demanding, i.e. phenol (G3). The behaviour of G3 in solution is very peculiar. In analogy to G2, the G3 molecule is unable to complex metal ions to form G4 stacked structures, as no changes can be detected both on CD (Fig. S4 and S8, ESI†) and on 1H NMR spectra after the addition of K+. Furthermore, in the absence of added ions, both N(1)–H and N(2)–H signals split upon cooling. In particular, the N(2)–H signal splits into two couples of new signals in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S5, ESI†). A couple of signals at ca. δ = 8 can be attributed to H-bonded N(2)–Hs, while the other couple of signals appearing at ca. δ = 3 ppm can be ascribed to free N(2)–Hs. The existence of two sets of resonances for both imino and amino protons in a 2:1 ratio points to the existence of two different supramolecular species. On the basis of NOE experiments (Fig. S6 and S7, ESI†), the major species can be ascribed to the formation of all-syn isolated G34. Although no direct and conclusive evidence could be gathered from the spectra, an in-depth inspection of the models suggests antiG34 or antiG3-dimers, analogous to those found for G2 on surfaces, as the possible structure for the minor species. STM investigation of sub-monolayer-thick films obtained from a (100 ± 2) μM solution of G3 revealed the formation of a new type of pattern. In this 2D crystal, because of steric hindrance brought into play by the phenol unit, only three out of four stearate side chains are physisorbed flat on the surface. The unit cell parameters amount to a = (4.5 ± 0.1) nm, b = (1.8 ± 0.1) nm, and α = (90 ± 2)°, leading to an area A = (8.1 ± 0.1) nm2, where each unit cell contains two molecules. Thus, the area occupied by a single molecule G3 corresponds to (4.1 ± 0.1) nm2. The packing of G3 molecules is very loose as evidenced by the large discrepancy between the areas occupied by single molecules G1, G2 and G3. The STM inset in Fig. 1e clearly shows the presence of macrocyclic bright features decorated with four small protrusions (indicated with white arrows in the inset), which can be assigned to G34 and ferrocene groups (backfolded into the supernatant solution), respectively. Because of the presence of sterically demanding phenol substituents in the C(8) position of G3, the formation of H-bonded ribbon-like structures is hindered,11 leading to the generation of cyclic tetrameric H-bonded structures characterized by the N(1)–H⋯O(6) and N(2)–H⋯N(7) motif, whose the existence was also indicated by NOE data in solution. While NOE analysis suggests the presence of all-syn isolated G34, as the main species, once adsorbed on the surface both all-syn and all-antiG34 will occupy the same areas, therefore we cannot unambiguously exclude the existence of the former over the latter. Noteworthily, some G34 appear brighter in the STM image, which can be explained by the interference of the supramolecular lattice and the underlying HOPG surface.
:1 ratio (Fig. S5, ESI†). A couple of signals at ca. δ = 8 can be attributed to H-bonded N(2)–Hs, while the other couple of signals appearing at ca. δ = 3 ppm can be ascribed to free N(2)–Hs. The existence of two sets of resonances for both imino and amino protons in a 2:1 ratio points to the existence of two different supramolecular species. On the basis of NOE experiments (Fig. S6 and S7, ESI†), the major species can be ascribed to the formation of all-syn isolated G34. Although no direct and conclusive evidence could be gathered from the spectra, an in-depth inspection of the models suggests antiG34 or antiG3-dimers, analogous to those found for G2 on surfaces, as the possible structure for the minor species. STM investigation of sub-monolayer-thick films obtained from a (100 ± 2) μM solution of G3 revealed the formation of a new type of pattern. In this 2D crystal, because of steric hindrance brought into play by the phenol unit, only three out of four stearate side chains are physisorbed flat on the surface. The unit cell parameters amount to a = (4.5 ± 0.1) nm, b = (1.8 ± 0.1) nm, and α = (90 ± 2)°, leading to an area A = (8.1 ± 0.1) nm2, where each unit cell contains two molecules. Thus, the area occupied by a single molecule G3 corresponds to (4.1 ± 0.1) nm2. The packing of G3 molecules is very loose as evidenced by the large discrepancy between the areas occupied by single molecules G1, G2 and G3. The STM inset in Fig. 1e clearly shows the presence of macrocyclic bright features decorated with four small protrusions (indicated with white arrows in the inset), which can be assigned to G34 and ferrocene groups (backfolded into the supernatant solution), respectively. Because of the presence of sterically demanding phenol substituents in the C(8) position of G3, the formation of H-bonded ribbon-like structures is hindered,11 leading to the generation of cyclic tetrameric H-bonded structures characterized by the N(1)–H⋯O(6) and N(2)–H⋯N(7) motif, whose the existence was also indicated by NOE data in solution. While NOE analysis suggests the presence of all-syn isolated G34, as the main species, once adsorbed on the surface both all-syn and all-antiG34 will occupy the same areas, therefore we cannot unambiguously exclude the existence of the former over the latter. Noteworthily, some G34 appear brighter in the STM image, which can be explained by the interference of the supramolecular lattice and the underlying HOPG surface.
To provide a molecular understanding of the self-assembly of the three G derivatives in 2D and gain insight into the formation and stability of supramolecular structures, we have performed density functional theory (DFT) calculations using the hybrid Gaussian and plane-wave method (GPW), implemented in the QUICKSTEP module of the CP2K package.14 We used the B3LYP hybrid exchange–correlation potential,15 whereas Grimme's DFT-D2 method16 was employed for taking into account the dispersion forces. The additional details of the computational methodology, as well as of the results for the structural and electronic properties of the different assembly motifs, are provided in the ESI.† To bestow information on the intermolecular binding mechanisms, we have focused our attention on unravelling the interplay between H-bonds, holding together the guanine cores and the effective metallic repulsion coming from the four iron cations present in the ferrocenes.
Noteworthily, as can be seen in the suggested monolayer packing motifs, two types of intramolecular interactions can be distinguished, i.e., the hydrogen-bonding (or N(2)–H⋯Br(8) interactions in the case of the G2 structure) between G cores and the van der Waals interaction, resulting from the interdigitation of the stearate chains. In order to determine their contribution to the total cohesive energy, we calculated the intermolecular dissociation energy for each of the different four-molecule-based configurations (see Fig. 2) exhibited in three G-based complexes, and the results are reported in Table 1. The presented first-principle calculations have not only elucidated the different mechanically stable molecular arrangements, but more importantly, have shed light on the energetics of intra-molecular (within unit cells) as well as inter-molecular (between neighbouring cells) interactions determining the stability of the molecular networks. According to Etot values, the ribbon structure of G1 is greatly stabilized by four strong H-bonds. In the G2 lamellar structure molecules are held together by two H-bonds to form dimers, which further self-assemble via two strong N(2)–H⋯Br bonds to form 1D arrays. As expected the G34 macrocycle is energetically unfavored, since the H-bonds involved in pairing are of weakest nature. Our findings indicate that the formation of intermolecular H-bonds guides the self-assembly, since the interactions between the stearate chains are much weaker. In the gas phase, the calculated electronic structure of the dimers and ribbons exhibits hybridization between the states stemming from the organic complex, namely the guanine backbone, and the metallic states associated with the ferrocene groups. The information given by the electronic structure of calculated complexes confirms that they are primarily held together by H-bonds even in the presence of the metallic repulsion coming from the occupied molecular orbitals with clear d symmetries.
| System | E tot | E H-bond | E Br–HN |
|---|---|---|---|
| a E tot, total intermolecular interaction energy, EH-bond, average hydrogen-bond energy, EBr–HN, energy of bromine–NH interactions, all energies in kcal mol−1. | |||
| G1 | −52.1 | −13.0 | — |
| G2 | −50.8 | −11.9 | −13.9 |
| G3 | −40.1 | −10.0 | — |
In summary, we have designed and synthesised novel organic soluble ferrocene-exposing guanosines. Their self-association in solutions, occurring via H-bonding, depends on the steric hindrance and H-bonding ability of the substituent attached to the nucleobase C(8)-position. When physisorbed at the solid/liquid interface the diversity of self-assembly behaviour upon chemical design is reflected in the generation of either different G-ribbon structures or the G4 cation-free architectures. These structures have been monitored on the sub-nm scale by in situ STM imaging. Our approach demonstrates that a careful molecular design of the guanosine starting building block makes it possible to steer the self-assembly towards the formation of different supramolecular architectures, even in the absence of templating ions. Such motifs are different, genuine supramolecular 2D scaffolds dictating, in the present case, the spatial localization of ferrocenes, ultimately forming 1D arrays that may be of interest in opto-electronics.
This work was supported by the European Community through the project EC FP7 ICT-MOLARNET (318516) and the European Research Council project SUPRAFUNCTION (GA-257305), the Agence Nationale de la Recherche through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC) and the International Center for Frontier Research in Chemistry (icFRC).
Notes and references
- (a) J. S. Lindsey, New J. Chem., 1991, 15, 153–180 CAS; (b) J.-M. Lehn, Supramolecular chemistry: concepts and perspectives, VCH, New York, 1995 Search PubMed; (c) G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. N. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS; (d) D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1155–1196 CrossRef CAS PubMed; (e) J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS; (f) D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403–2407 CrossRef CAS PubMed; (g) J. F. Stoddart and H. R. Tseng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797–4800 CrossRef CAS PubMed.
- (a) G. Gottarelli, S. Masiero, E. Mezzina, S. Pieraccini, J. P. Rabe, P. Samorì and G. P. Spada, Chem. – Eur. J., 2000, 6, 3242–3248 CrossRef CAS; (b) S. Lena, G. Brancolini, G. Gottarelli, P. Mariani, S. Masiero, A. Venturini, V. Palermo, O. Pandoli, S. Pieraccini, P. Samorì and G. P. Spada, Chem. – Eur. J., 2007, 13, 3757–3764 CrossRef CAS PubMed; (c) A. Ciesielski, S. Lena, S. Masiero, G. P. Spada and P. Samorì, Angew. Chem., Int. Ed., 2010, 49, 1963–1966 CrossRef CAS PubMed; (d) A. Ciesielski, R. Perone, S. Pieraccini, G. P. Spada and P. Samorì, Chem. Commun., 2010, 46, 4493–4495 RSC.
- (a) M. Yu, J. G. Wang, M. Mura, Q. Q. Meng, W. Xu, H. Gersen, E. Laegsgaard, I. Stensgaard, R. E. A. Kelly, J. Kjems, T. R. Linderoth, L. N. Kantorovich and F. Besenbacher, ACS Nano, 2011, 5, 6651–6660 CrossRef CAS PubMed; (b) A. Ciesielski, S. Haar, A. Benyei, G. Paragi, C. F. Guerra, F. M. Bickelhaupt, S. Masiero, J. Szolomajer, P. Samori, G. P. Spada and L. Kovacs, Langmuir, 2013, 29, 7283–7290 CrossRef CAS PubMed; (c) A. Ciesielski, S. Haar, G. Paragi, Z. Kupihar, Z. Kele, S. Masiero, C. F. Guerra, F. M. Bickelhaupt, G. P. Spada, L. Kovacs and P. Samori, Phys. Chem. Chem. Phys., 2013, 15, 12442–12446 RSC.
- (a) G. P. Spada, S. Lena, S. Masiero, S. Pieraccini, M. Surin and P. Samorì, Adv. Mater., 2008, 20, 2433–2438 CrossRef CAS PubMed; (b) T. F. A. Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef PubMed.
- Y. L. Wu, K. E. Brown and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 13322–13325 CrossRef CAS PubMed.
- (a) J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed; (b) J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- J. L. Sessler and R. Z. Wang, Angew. Chem., Int. Ed., 1998, 37, 1726–1729 CrossRef CAS.
- V. Andrisano, G. Gottarelli, S. Masiero, E. H. Heijne, S. Pieraccini and G. P. Spada, Angew. Chem., Int. Ed., 1999, 38, 2386–2388 CrossRef CAS.
- G. Gottarelli, S. Masiero, E. Mezzina, G. P. Spada, P. Mariani and M. Recanatini, Helv. Chim. Acta, 1998, 81, 2078–2092 CrossRef CAS.
- (a) J. A. Walmsley and J. F. Burnett, Biochemistry, 1999, 38, 14063–14068 CrossRef CAS PubMed; (b) T. Giorgi, S. Lena, P. Mariani, M. A. Cremonini, S. Masiero, S. Pieraccini, J. P. Rabe, P. Samorì, G. P. Spada and G. Gottarelli, J. Am. Chem. Soc., 2003, 125, 14741–14749 CrossRef CAS PubMed.
- J. L. Sessler, M. Sathiosatham, K. Doerr, V. Lynch and K. A. Abboud, Angew. Chem., Int. Ed., 2000, 39, 1300–1303 CrossRef CAS.
- (a) R. Otero, M. Schock, L. M. Molina, E. Laegsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Angew. Chem., Int. Ed., 2005, 44, 2270–2275 CrossRef CAS PubMed; (b) R. Otero, W. Xu, M. Lukas, R. E. A. Kelly, E. Laegsgaard, I. Stensgaard, J. Kjems, L. N. Kantorovich and F. Besenbacher, Angew. Chem., Int. Ed., 2008, 47, 9673–9676 CrossRef CAS PubMed.
- A. Ciesielski and P. Samorì, Nanoscale, 2011, 3, 1397–1410 RSC.
- (a) M. Krack and M. Parrinello, High Performance Computing in Chemistry, 2004, vol. 25, pp. 29–51 Search PubMed; (b) J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis, full characterisation of new compounds, NMR spectra, and DFT. See DOI: 10.1039/c5cc03197d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |