
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-catalyzed ortho alkenylation of aromatic nitriles with activated alkenes via C–H bond activation†
Mallu Chenna
Reddy
and
Masilamani
Jeganmohan
*
Department of Chemistry, Indian Institute of Science Education and Research, Pune 411021, India. E-mail: mjeganmohan@iiserpune.ac.in
First published on 21st May 2015
Abstract
The ruthenium-catalyzed ortho alkenylation of substituted aromatic and heteroaromatic nitriles with activated alkenes providing ortho alkenylated aromatic and heteroaromatic nitriles in a highly regio- and stereoselective manner is described.
Selective transformation of the C–H bond of organic moieties into C–C and C–heteroatom bond catalyzed by transition metal complexes via C–H bond activation is one of the most versatile and well-acknowledged methods in organic synthesis.1 This transformation has gained tremendous attention in chemical and pharmaceutical industries, because it provides step- and atom-economical routes to synthesize useful organic molecules from the readily available starting materials.2 Particularly, a transition metal-catalyzed oxidative cross-coupling of heteroatom substituted aromatics with alkenes has proven to be a highly efficient route to synthesize disubstituted alkenes without having any prefunctionalized starting materials in a highly regio- and stereoselective manner.3 The selectivity of the C–H bond of organic moieties can be controlled by using suitable directing groups. In most cases, the lone pair of electrons of nitrogen or oxygen atom of the directing groups coordinate with the metal complex through σ-coordination and activate the C–H bond of organic moieties selectively (Fig. 1, eqn (1)).4,5 The search for new variants to activate the C–H bond of aromatics is very important to expand the synthetic scope of the alkenylation reaction. Very recently, Cheng's group disclosed an alkene assisted alkenylation of aromatics with activated alkenes in the presence of a palladium catalyst via an unusual carbon–carbon π-bond coordination (Fig. 1, eqn (2)).6
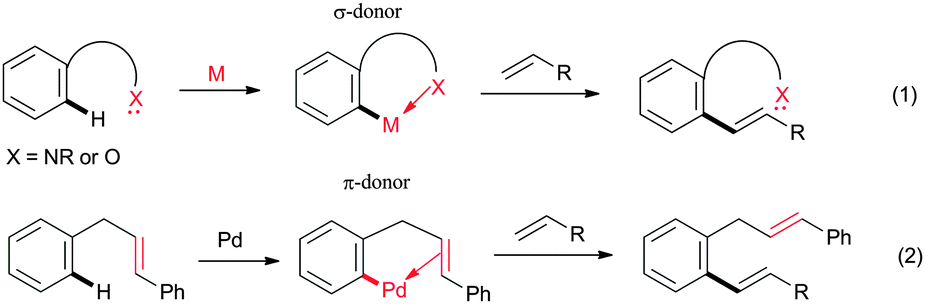 | ||
| Fig. 1 Chelation-assisted C–H bond alkenylation. | ||
Nitrile is a versatile functional group which can be efficiently used for various organic transformations.7 In addition, nitrile group containing organic molecules are used as pharmaceuticals, pesticides and dyes.8 The strong electron withdrawing nature and better hydrogen bond accepting properties of the nitrile group allow it to be used widely in designing drug molecules. Meanwhile, the cyano group can also be used as a directing group for the C–H bond activation reactions.9–11 It is known that the lone pair of electrons of the nitrogen atom of the nitrile group of benzonitrile coordinate with the metal complex through σ-coordination, and this process leads to the formation of a linear metal complex.9 Also, a less likely, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N π-bond of benzonitrile coordinates with the metal, yielding the π-coordinated metal complexes.10 Recently, by employing the σ-coordination of the nitrile moiety, the meta C–H bond of aromatics could be activated efficiently for the alkenylation reaction (Fig. 2, eqn (1)).11 To date, there is no report on the CN π-bond assisted alkenylation at the ortho position of aromatic nitriles with alkenes due to the difficulty in coordination of the π-bond of CN with metals.
N π-bond of benzonitrile coordinates with the metal, yielding the π-coordinated metal complexes.10 Recently, by employing the σ-coordination of the nitrile moiety, the meta C–H bond of aromatics could be activated efficiently for the alkenylation reaction (Fig. 2, eqn (1)).11 To date, there is no report on the CN π-bond assisted alkenylation at the ortho position of aromatic nitriles with alkenes due to the difficulty in coordination of the π-bond of CN with metals.
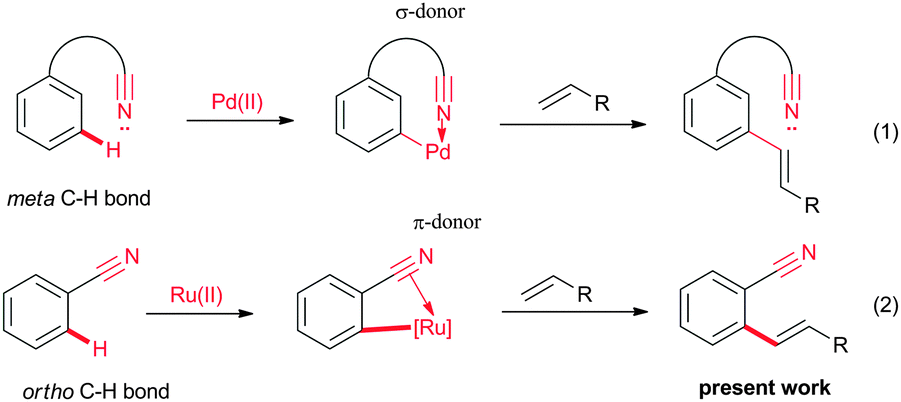 | ||
| Fig. 2 C–H bond alkenylation of aromatic nitriles. | ||
Our ongoing interest in the finding of a new C–H bond transformation reaction prompted us to explore the possibility of CN π-bond assisted ortho alkenylation of substituted aromatic nitriles with alkenes. Herein, we wish to report for the first time nitrile as a π-bond coordinating group for the ortho alkenylation of aromatic and heteroaromatic nitriles with activated alkenes in the presence of a ruthenium catalyst (Fig. 2, eqn (2)). The alkenylation reaction was compatible with functional group substituted aromatic nitriles. Later, the ortho alkenylated aromatic nitrile was converted into a chiral phthalide in the presence of AD-mix-β. By employing nitrile as a directing group, arylation was performed at the alkene C–H bond of the ortho alkenylated aromatic nitriles with aromatic iodides in the presence of a Pd catalyst.
Initially, the ruthenium-catalyzed alkenylation reaction was examined using benzonitrile (1a) and n-butyl acrylate (2a) (Scheme 1). It is expected that the nitrogen atom of 1a prefers to coordinate with the metal via the lone pair electrons rather than π-coordination. However, for successful ortho C–H bond activation, π-coordination of nitrile is crucial. Meanwhile, a metal acetate base is needed for the deprotonation of the C–H bond of weak coordinating group substituted aromatics. Thus, a combination of the ruthenium catalyst and metal acetate having a Lewis acidic nature was selected. The main idea behind the selection of the Lewis acidic metal acetate is that the lone pair of nitrogen of the nitrile moiety would coordinate with the Lewis acid prior to the ruthenium catalyst reaction and block the corresponding site. Therefore, a possibility is created for the π-bond coordination of the nitrile moiety with the ruthenium catalyst and initiates the C–H bond activation. With this idea, a combination of [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and Cu(OAc)2·H2O (2.0 equiv.) was used for the reaction. As a nitrile is a weak coordinating group, AgSbF6 was used to generate a cationic ruthenium species for the C–H bond reaction. However, in the reaction, hydration takes place at the nitrile group and only benzamide (3a) was observed. If the same reaction is run for a long time, a cyclic isoindolin-1-one derivative was observed.12 Later, the reaction was examined using various Lewis acids such as Co(OAc)2, Mn(OAc)2, Ag2O, AgOAc, Ag2CO3, Ag(CF3CO2) and Fe(OAc)2 (2.0 equiv.). Very interestingly, in AgOAc, the ortho C–H bond activation takes place selectively and further reaction with n-butyl acrylate (2a) provides the ortho alkenylated benzonitrile 4aa in 75% isolated yield. In the reaction, benzamide 3a was observed only in a very minor 3% yield. In other silver salts such as Ag2O, Ag2CO3 and Ag(CF3CO2), the product 4aa was observed in 45%, 49% and 61% yields. Benzamide 3a was observed in 25%, 19% and 14% yields, respectively. But, in Co(OAc)2 and Mn(OAc)2, benzamide 3a was observed in more than 45% and 51% yields and product 4aa was observed in 15% and 19% yields, respectively. Meanwhile, the acidic solvent AcOH is crucial for the reaction. Other solvents such as 1,2-dichloroethane, THF, 1,4-dioxane, DMF, toluene and CF3COOH were not suitable for the reaction. However, in iso-PrOH, the product 4aa was observed only in 10% yield without benzamide 3a formation.
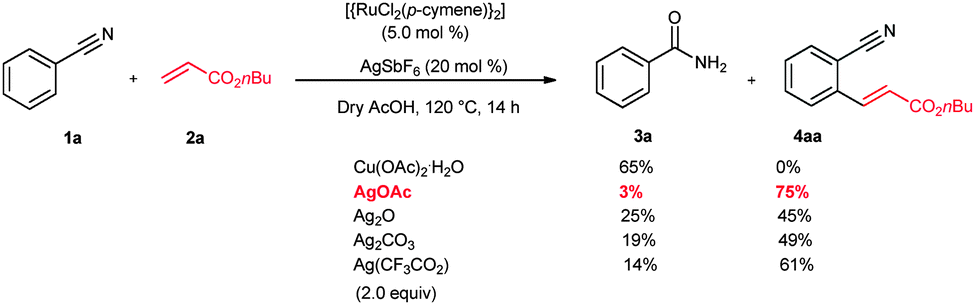 | ||
| Scheme 1 ortho Alkenylation of benzonitrile. | ||
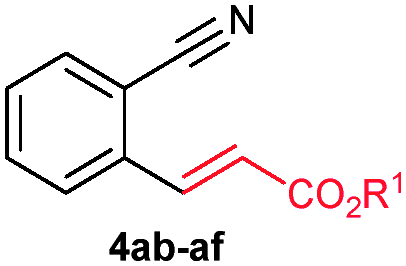
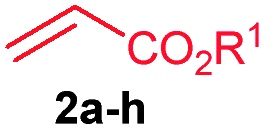
The scope of the alkenylation reaction was examined with various activated alkenes 2 under the optimized reaction conditions (Table 1). Ethyl acrylate (2b), cyclohexyl acrylate (2c), phenyl acrylate (2d), benzyl acrylate (2e) and 2-phenoxyethyl acrylate (2f) reacted efficiently with 1a, providing the expected ortho alkenylated benzonitriles 4ab–af in 64%, 79%, 69%, 67% and 59% yields, respectively (entries 1–5). Further, n-butyl acrylate (2a), ethyl acrylate (2b), methyl acrylate (2g) and 2,2,2-trifluoroethyl acrylate (2h) also efficiently reacted with 2-naphthonitrile (1b), yielding ortho alkenylated 2-naphthonitriles 4ba–bh in 75%, 65%, 62% and 57% yields, respectively (entries 6–9). In the reaction, C–H bond activation takes place selectively at the C3 position of 2-naphthonitrile (1b). Interestingly, phenyl vinyl sulphone (2i) was also efficiently involved in the reaction, providing product 4bi in 52% yield (entry 10).
| Entry | Alkenes 2 | Product 4 | Yieldb (%) |
|---|---|---|---|
| a All the reactions were carried out using 1a or 1b (75 mg), alkenes 2a–h (4.0 equiv.) and 2i (2.0 equiv.), [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and AgOAc (2.0 equiv.) in dry acetic acid (3.0 mL) at 120 °C for 14 h. b Isolated yield. c In this reaction, DCE (3.0 mL) + pivalic acid (0.5 mL) was used instead of AcOH. | |||
|
|
||
| 1 | 2b: R1 = Et | 4ab: R1 = Et | 64 |
| 2 | 2c: R1 = cyclohexyl | 4ac: R1 = cyclohexyl | 79 |
| 3 | 2d: R1 = Ph | 4ad: R1 = Ph | 69 |
| 4 | 2e: R1 = CH2Ph | 4ae: R1 = CH2Ph | 67 |
| 5 | 2f: R1 = (CH2)2OPh | 4af: R1 = (CH2)2OPh | 59 |
|

|
||
| 6 | 2a: R1 = n-Bu | 4ba: R1 = n-Bu | 75 |
| 7 | 2b: R1 = Et | 4bb: R1 = Et | 65 |
| 8 | 2g: R1 = Me | 4bg: R1 = Me | 62 |
| 9 | 2h: R1 = CH2CF3 | 4bh: R1 = CH2CF3 | 57 |
| 10 |

|

|
52c |
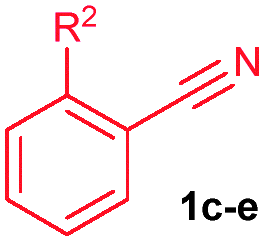
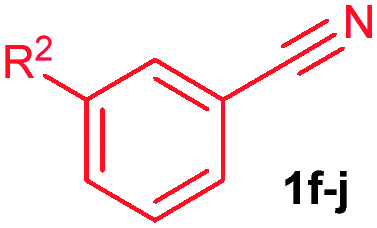
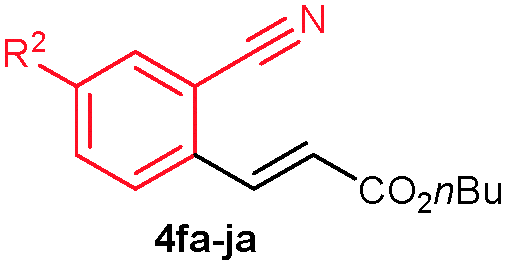
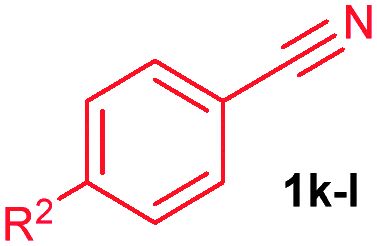
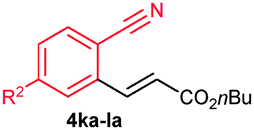
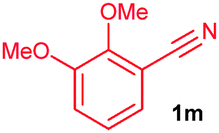
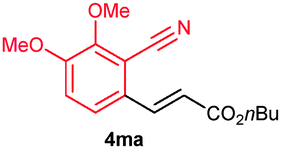
The alkenylation reaction was compatible for a variety of sensitive functional groups such as Br, Cl, I, OMe, SMe and CO2Me substituted aromatic nitriles (Table 2). ortho Bromo (1c), chloro (1d) and methoxy (1e) substituted benzonitriles reacted efficiently with n-butyl acrylate (2a), providing the corresponding ortho alkenylated aromatic nitriles 4ca–ea in 51%, 50% and 47% yields, respectively (entries 1–3). Subsequently, meta methylester (1f), iodo (1g), bromo (1h), chloro (1i) and methoxy (1j) benzonitriles also efficiently participated in the reaction, providing the corresponding ortho alkenylated benzonitriles 4fa–ja in 53%, 48%, 45%, 44% and 42% yields, respectively, in a highly regioselective manner (entries 4–8). In all these reactions, C–H bond activation takes place at a less hindered ortho C6–H bond of the benzonitriles (1f–j). 4-Methoxy (1k) and 4-SMe (1l) substituted benzonitriles provided the corresponding alkenylation products 4ka and 4la in 42% and 57% yields, respectively (entries 9 and 10). The alkenylation reaction was also examined with disubstituted benzonitriles (1m–o). 2,3-Dimethoxy (1m) and 3,4-dimethoxy (1n) benzonitriles reacted with 2a or 2b, affording ortho alkenylated benzonitriles 4ma and 4nb in moderate 38% and 29% yields, respectively (entries 11 and 12). In the substrate 1n, the C–H bond activation takes place at the less hindered ortho C6–H bond. In contrast, a sterically hindered ortho C–H bond of piperonylonitrile (1o) reacted with 2a or 2g providing products 4oa and 4og in 52% and 48% yields, respectively (entries 13 and 14). The present result clearly reveals that a strong electron donating OMe group as the substituent on the benzonitrile decreases the yield of the product, and SMe, halogens and electron withdrawing substituents on the benzonitrile moderately increases the yield. In unsubstituted benzonitrile and 2-naphthonitrile substrates, good yields were observed.
| Entry | Alkenes 2 | Product 4 | Yieldb (%) |
|---|---|---|---|
| a All the reactions were carried out using 1c–o (75 mg), alkenes 2 (4.0 equiv.), [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and AgOAc (2.0 equiv.) in dry acetic acid (3.0 mL) at 120 °C for 14 h. b Isolated yield. c The reaction time was 16 h. | |||
|

|
||
| 1 | 1c: R2 = Br | 4ca: R2 = Br | 51 |
| 2 | 1d: R2 = Cl | 4da: R2 = Cl | 50 |
| 3 | 1e: R2 = OMe | 4ea: R2 = OMe | 47 |
|
|
||
| 4 | 1f: R2 = CO2Me | 4fa: R2 = CO2Me | 53 |
| 5 | 1g: R2 = I | 4ga: R2 = I | 48 |
| 6 | 1h: R2 = Br | 4ha: R2 = Br | 45 |
| 7 | 1i: R2 = Cl | 4ia: R2 = Cl | 44 |
| 8 | 1j: R2 = OMe | 4ja: R2 = OMe | 42 |
|
|
||
| 9 | 1k: R2 = OMe | 4ka: R2 = OMe | 42c |
| 10 | 1l: R2 = SMe | 4la: R2 = SMe | 57 |
| 11 |
|
|
38c |
| 12 |

|

|
29c |

|

|
||
| 13 | 4oa: R2 = n-Bu | 52c | |
| 14 | 4og: R2 = Me | 48c | |
The alkenylation reaction was also successfully extended with heteroaromatic nitriles (Scheme 2). Treatment of 2-nitrile thiophene (1p) with n-butyl acrylate (2a) in the presence of [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and AgOAc (2.0 equiv.) at 120 °C for 16 h provided 2-alkenyl-3-nitrile thiophene (4pa) in 53% yield. 3-Nitrile thiophene (1q) reacted with n-butyl acrylate (2a) affording bis alkenylated 3-nitrile thiophene 5qa in 25% yield. Next, the same reaction was examined using Cu(OAc)2·H2O without AgOAc. Interestingly, in this reaction, bis alkenylated 3-nitrile thiophene 5qa was observed in 57% yield (Scheme 3). Similarly, n-ethyl acrylate (2b), cyclohexyl acrylate (2c), phenyl acrylate (2d), benzyl acrylate (2e), 2-phenoxyethyl acrylate (2f) and methyl acrylate (2g) also reacted with 1q, affording bis alkenylated 3-nitrile thiophenes 5qb–qg in 65%, 48%, 49%, 47%, 45% and 69% yields, respectively (Scheme 3).
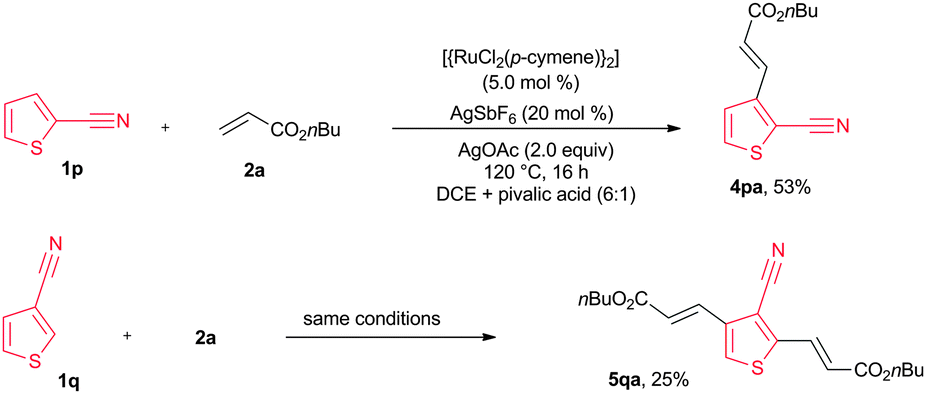 | ||
| Scheme 2 Alkenylation of 2-cyano and 3-cyanothiophenes. | ||
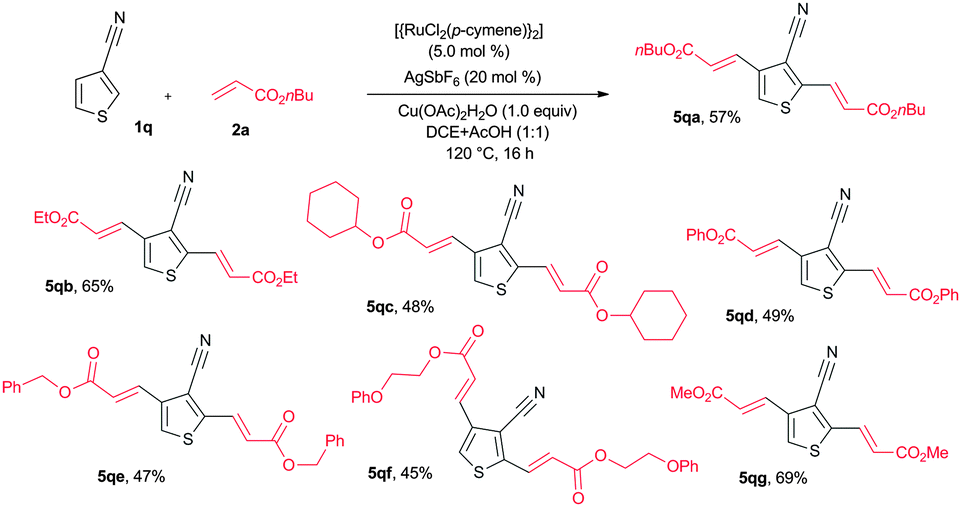 | ||
| Scheme 3 bis Alkenylation of 3-cyanothiophene. | ||
A substituted ortho alkenyl aromatic nitrile is a versatile synthetic intermediate which can be used for synthesizing various useful organic molecules.7,8ortho Alkenyl benzonitrile 4ab underwent an intramolecular cyclization in the presence of AD-mix-β, yielding a chiral phthalide 6 in 93% yield in 99 ee% (Scheme 4).13 By using AD-mix-α, the reverse chiral phthalide derivative can be prepared in a highly enantioselective manner.13 Further, by employing nitrile as a directing group, arylation was performed at the alkene C–H bond of 4ab and 4aa with 4-iodo nitrobenzene (7) in the presence of Pd(OAc)2 and Ag2O in CF3COOH at 110 °C for 12 h, giving trisubstituted alkenes 8a and 8b in 82% and 87% yields in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z:E ratio.
:1 Z:E ratio.
 | ||
| Scheme 4 Transformation of ortho alkenyl benzonitriles 4. | ||
A possible reaction mechanism is proposed to account for the present alkenylation reaction in Scheme 5. It is strongly believed that first the lone electron pair of the nitrogen atom of benzonitrile 1 coordinates with the Lewis acid AgOAc, providing a linear benzonitrile silver complex 9. A similar observation is strongly supported by DFT calculations.14 Subsequently, AgSbF6 likely removes the Cl− ligand from the [{RuCl2(p-cymene)}2] complex, providing a cationic ruthenium species 10. Coordination of the CN π-bond of 9 into the ruthenium species10 followed by ortho-metalation provides intermediate 11. Coordinative insertion of activated alkene 2 into the Ru–carbon bond of intermediate 11 affords intermediate 12. Subsequent β-hydride elimination of intermediate 12 in the presence of AgOAc gives product 4 and regenerates the active ruthenium species 10. To support the hypothesis that ortho C–H bond cleavage of 9 is a reversible and rate determining process, the reaction of 1b was carried out in the presence of CD3COOD under similar reaction conditions. In this reaction, the product D-1b was observed in 40% yield, in which 40% of deuterium incorporation was observed at both C-1 as well as C-3 carbons of 1b.
 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, we have demonstrated a ruthenium-catalyzed CN π-bond assisted ortho alkenylation of substituted aromatic and heteroaromatic nitriles with activated alkenes providing ortho alkenylated aromatic and heteroaromatic nitriles in good to moderate yields in a highly regio- and stereoselective manner.
We thank the CSIR (02(0179)/14/EMR-II), India, for the support of this research. M.C.R. thanks the CSIR for a fellowship.
Notes and references
- Selected references: (a) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (e) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (f) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- Selected papers: (a) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed; (b) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- (a) J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- (a) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970 CrossRef CAS PubMed; (b) T. Ueyama, S. Mochida, T. Fukutani, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 706 CrossRef CAS PubMed; (c) K. Padala, S. Pimparkar, P. Madasamy and M. Jeganmohan, Chem. Commun., 2012, 48, 7140 RSC; (d) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef CAS PubMed; (e) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (f) D. Zhao, C. Nimphius, M. Lindale and F. Glorius, Org. Lett., 2013, 15, 4504 CrossRef CAS PubMed; (g) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134 CrossRef CAS PubMed; (h) W. Ma, R. Mei, G. Tenti and L. Ackermann, Chem. – Eur. J., 2014, 20, 15248 CrossRef CAS PubMed; (i) W. Ma and L. Ackermann, Chem. – Eur. J., 2013, 19, 13925 CrossRef CAS PubMed; (j) M. C. Reddy and M. Jeganmohan, Eur. J. Org. Chem., 2013, 1150 CrossRef CAS PubMed; (k) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS PubMed; (l) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed.
- (a) H.-L. Wang, R.-B. Hu, H. Zhang, A.-X. Zhou and S.-D. Yang, Org. Lett., 2013, 15, 5302 CrossRef CAS PubMed; (b) K. Parthasarathy and C. Bolm, Chem. – Eur. J., 2014, 20, 4896 CrossRef CAS PubMed; (c) Y. Yokoyama, Y. Unoh, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 7649 CrossRef CAS PubMed; (d) F. W. Patureau, T. Besset and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1064 CrossRef CAS PubMed; (e) K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169 CrossRef CAS PubMed; (f) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242 CrossRef CAS PubMed; (g) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075 RSC; (h) C. Wang and H. B. Ge, Chem. – Eur. J., 2011, 17, 14371 CrossRef CAS PubMed; (i) B. Li, K. Devaraj, C. Darcel and P. H. Dixneuf, Green Chem., 2012, 14, 2706 RSC; (j) K. S. Singh and P. H. Dixneuf, Organometallics, 2012, 31, 7320 CrossRef CAS; (k) K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541 CrossRef CAS PubMed; (l) W. Dong, K. Parthasarathy, Y. Cheng, F. Pan and C. Bolm, Chem. – Eur. J., 2014, 20, 15732 CrossRef CAS PubMed.
- (a) P. Gandeepan and C.-H. Cheng, Chem. – Asian J., 2015, 10, 824 CrossRef CAS PubMed; (b) P. Gandeepan and C.-H. Cheng, J. Am. Chem. Soc., 2012, 134, 5738 CrossRef CAS PubMed.
- (a) Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers, London, 1970 Search PubMed; (b) R. C. Larock, Comprehensive OrganicTransformations: A Guide to Functional Group Preparations, VCH, NewYork, 1989 Search PubMed; (c) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC.
- (a) K. Friedrich and K. Wallenfels, in The Chemistry of the Cyano Group, ed. Z. Rappaport, Wiley, New York, 1970 Search PubMed; (b) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed.
- (a) J. A. Davies and F. R. Hartley, Chem. Rev., 1981, 81, 79 CrossRef CAS; (b) R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299 CrossRef CAS; (c) M. F. Farona and N. J. Bremer, J. Am. Chem. Soc., 1966, 88, 3735 CrossRef CAS.
- (a) F. Kakiuchi, M. Sonoda, T. Tsujimoto, N. Chatani and S. Murai, Chem. Lett., 1999, 1083 CrossRef CAS; (b) W. Li, Z. Xu, P. Sun, X. Jiang and M. Fang, Org. Lett., 2011, 13, 1286 CrossRef CAS PubMed; (c) J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742 CrossRef CAS PubMed; (d) W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362 CrossRef CAS PubMed; (e) B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786 CrossRef CAS PubMed.
- (a) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed; (b) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS PubMed; (c) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760 CrossRef CAS PubMed.
- M. C. Reddy and M. Jeganmohan, Org. Lett., 2014, 16, 4866 CrossRef CAS PubMed.
- R. S. Reddy, I. N. C. Kiran and A. Sudalai, Org. Biomol. Chem., 2012, 10, 3655 CAS.
- T. Shoeib, H. E. Aribi, K. W. M. Siu and A. C. Hopkinson, J. Phys. Chem. A, 2001, 105, 710 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data. See DOI: 10.1039/c5cc03112e |
| This journal is © The Royal Society of Chemistry 2015 |