
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electron-conducting pyrene-fused phenazinothiadiazole†
A. Belen
Marco
a,
Diego
Cortizo-Lacalle
a,
Cristian
Gozalvez
a,
Mikel
Olano
a,
Ainhoa
Atxabal
b,
Xiangnan
Sun
*b,
Manuel
Melle-Franco
c,
Luis E.
Hueso
 bd and
Aurelio
Mateo-Alonso
*ad
bd and
Aurelio
Mateo-Alonso
*ad
aPOLYMAT, University of the Basque Country UPV/EHU, Avenida de Tolosa 72, E-20018 Donostia-San Sebastian, Spain. E-mail: amateo@polymat.eu
bCIC nanoGUNE Consolider, Avenida de Tolosa 76, E-20018 Donostia-San Sebastian, Spain. E-mail: x.sun@nanogune.eu
cCentro ALGORITMI, 4710-057 Braga, Portugal
dIkerbasque, Basque Foundation for Science, Bilbao, Spain
First published on 22nd May 2015
Abstract
A pyrene-fused phenazinothiadiazole that shows electron mobilities (μe = 0.016 cm2 V−1 s−1) two orders of magnitude higher than those reported for pyrene-fused pyrazaacenes is described.
Advances in organic electronics are expected to arise by gaining control over the electronic properties of molecular materials and their organisation in thin films.1 For instance, charge transport on molecular materials depends both on the molecular structure that provides distinct electronic properties, and also on the nature of the interactions between adjacent molecules that give rise to different structures and morphologies at the nanoscale. In these terms, pyrene-fused pyrazaacenes2 (PPAs) have recently re-emerged as a platform for developing novel organic semiconductors because of their tunable electronic structure,3 enhanced stability,4 and ability to self-organise into discotic phases5 and complex nanostructures.6 Furthermore, PPAs combines p-type pyrene and n-type azaacene residues in their structure, and therefore they can transport holes or electrons depending on their electronic structure. While hole mobilities7 up to 10−2 cm2 V−1 s−1 have been reported for PPA, the conduction of electrons still remains a significant challenge. In fact, mobilities in the range of 10−4 cm2 V−1 s−1 have been reported for electrons.8
In this work, we describe the synthesis and characterisation of pyrene-fused phenazinothiadiazole 1 (Scheme 1) that shows electron mobilities (μe = 0.016 cm2 V−1 s−1) two orders of magnitude higher than those reported for PPA. Such enhanced electron mobilities are consistent with an optimal LUMO level alignment, a favourable molecular packing and the large aspect ratio of the self-assembled nanostructures obtained upon deposition.
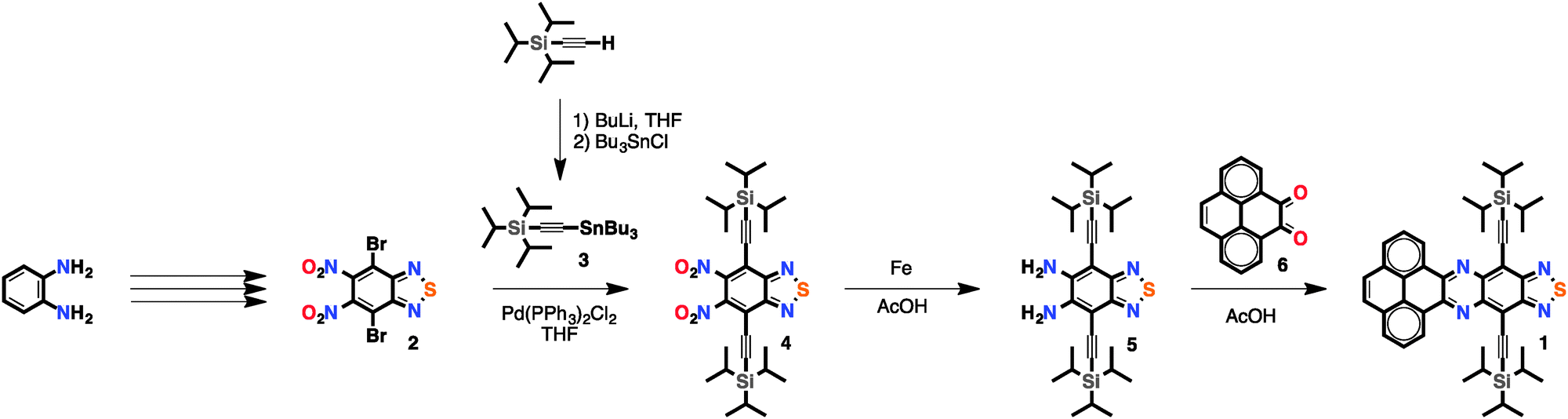 | ||
| Scheme 1 Synthesis of pyrene-fused phenazinothiadiazole 1. | ||
Pyrene-fused phenazinothiadiazole 1 was prepared following the synthetic route set out on Scheme 1. Compound 29 was synthesised in three steps from o-phenylenediamine and then transformed into 4 by Stille coupling using acetylene 3 that gave almost identical results compared to a similar procedure10 with trimetyltin acetylene. Compound 3 was prepared by lithiation of the commercially available (triisopropylsilyl)acetylene followed by nucleophilic substitution with tributyltin chloride.11 Diamine 5 was obtained upon the reduction of the nitro groups of 4 with Fe in acetic acid. Pyrene-fused phenazinothiadiazole 1 was obtained by cyclocondensation of 5 with pyrene diketone 612 in acetic acid at 80 °C. Compound 1 is a purple solid that is readily soluble in a wide variety of organic solvents including CH2Cl2, CHCl3, THF and o-dichlorobenzene (ODCB).
Single crystals of pyrene-fused phenazinothiadiazole 1 were grown to investigate the underlying molecular structure and solid-state packing. Crystals suitable for X-ray diffraction were obtained by slow evaporation from CH2Cl2 solutions. The crystal structure of 1 (Fig. 1) confirms the molecular structure and provides an additional insight into the electronic structure. The aromatic framework of 1 is essentially flat while the ethylene substituents are slightly out of plane. The bond lengths are consistent with the presence of Clar sextets on rings A and B (∼140 pm), and a localised double bond on ring C (135 pm). The bonds connecting the phenanthrene residue with the diazonaphthothiadiazole residue are longer than a standard aromatic bond (147 pm), which is consistent with a smaller electron density on D and thus with Clar rules. The diazonaphthothiadiazole residue presents approximately the expected bond lengths for pyrazine (ring E), benzene (ring F) and thiadiazol (ring G). Pyrene-fused phenazinothiadiazole 1 molecules pile-up with their aromatic moieties in an anti-parallel fashion so that the aromatic rings sit snugly between the bulky TIPS groups of their two neighbours at virtually the interlayer graphite distance. There are two distances at 3.35 Å and another at 3.40 Å. The existing face-to-face packing provides a pathway for charge transport and suggests that 1 can in principle function as a semiconductor.
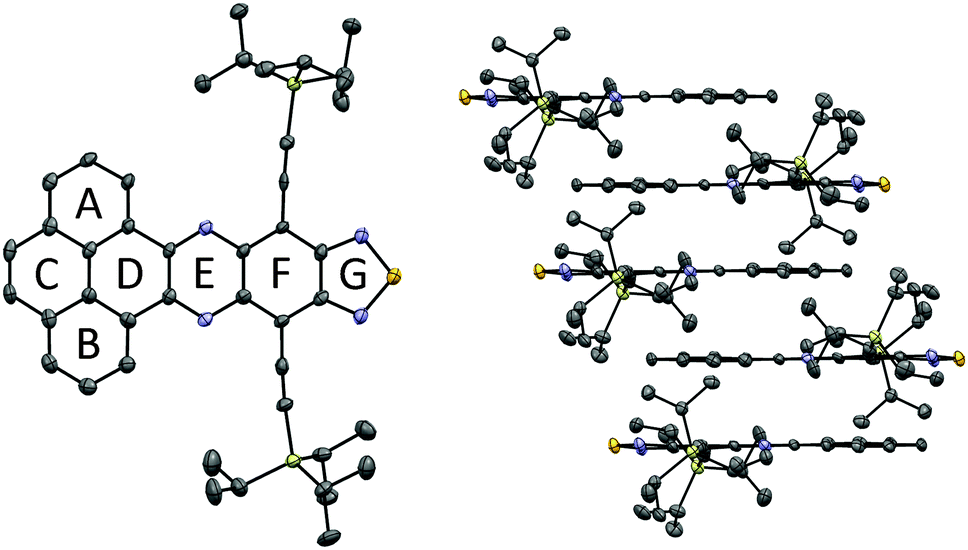 | ||
| Fig. 1 X-ray structure (ellipsoids at a 50% probability level) of pyrene-fused phenazinothiadiazole 1. C: grey, N: blue, S: orange, Si: yellow. | ||
The absorption spectrum of 1 (Fig. 2) in ODCB displays a broad absorption band in the visible region centred at 557 nm (log ε = 4.66) with resolved features with maxima at 525 (log ε = 4.66) and 599 (log ε = 451) nm. Also, a well-resolved absorption band is discernible in the UV region between 300 and 375 nm. The HOMO–LUMO gap was estimated from the absorption onset and corresponds to 1.96 eV (Table 1). The photoluminescence spectrum of 1 in ODCB obtained by excitation at 590 nm reveals a featureless band centred at 626 nm with a shoulder at 665 nm.
| λ max | λ em | E I1/2 | E II1/2 | E gap | E LUMO | E gap | E LUMO |
|---|---|---|---|---|---|---|---|
| a Measured in ODCB (nm). b Measured in 0.1 M nBu4PF6/ODCB (V), Ag wire pseudoreference electrode referenced vs. SCE using ferrocene (Fc) as the internal standard (EFc(SCE)1/2 = +0.48 V). c Estimated from the absorption onset (eV). d Estimated from CV EONSET (eV) according to ELUMO = −4.8 −e(EONSET − EFc1/2) where EFc1/2 was measured in situ. e Calculated B3LYP-ODBC-6311g+(d2,p)/B3LYP-ODBC-631(d,p). | |||||||
| 557 | 626 | −0.57 | −1.07 | 1.96 | −3.83 | 2.2 | −3.78 |
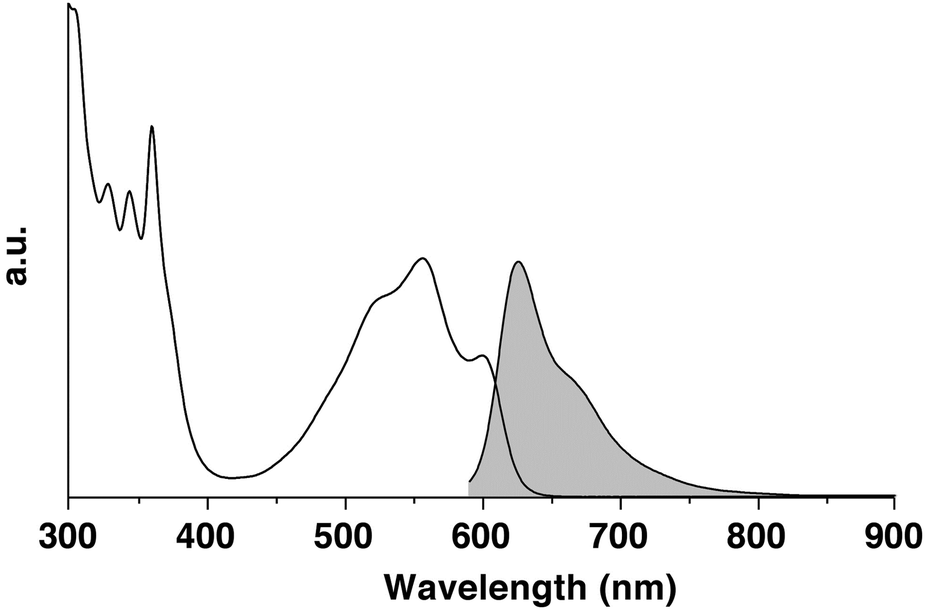 | ||
| Fig. 2 Absorption (white) and photoluminescence (grey, λexc = 590 nm) spectra of pyrene-fused phenazinothiadiazole 1. | ||
To shed light on the nature of the electronic transitions in the electronic spectrum of 1, the lower energy singlet excited states were calculated using the program Gaussian 0913 and using time-dependent density functional theory (TD-DFT B3LYP-ODBC-6311g+(d2,p)/B3LYP-ODBC-631(d,p)). The TD-DFT model (Table S4 and Fig. S11, ESI†) computes the experimental band centred at 557 nm as the sum of two electronic transitions: a HOMO−1 → LUMO transition at 613 nm and a weaker HOMO → LUMO transition at 683 nm with oscillator strengths of 0.30 and 0.23 respectively. In addition, the HOMO−2 → LUMO transition at 493 nm is allowed yet does not contribute notably for the simulated spectrum as it possesses a low oscillator strength: 0.02.
Cyclic voltammograms of 1 (0.1 M nBu4PF6 in ODCB) exhibit two fully-reversible reduction waves at E1/2 = −0.57 and −1.07 V (Fig. 3), which were assigned to the radical-anion and the dianion in analogy to phenazine derivatives.14 On the oxidative scan, no redox processes were observed within the solvent-supported electrolyte window. The electrochemical LUMO (or electron affinity) of 1 (−3.83 eV) was estimated from the potential onsets of the first reduction waves (Table 1).
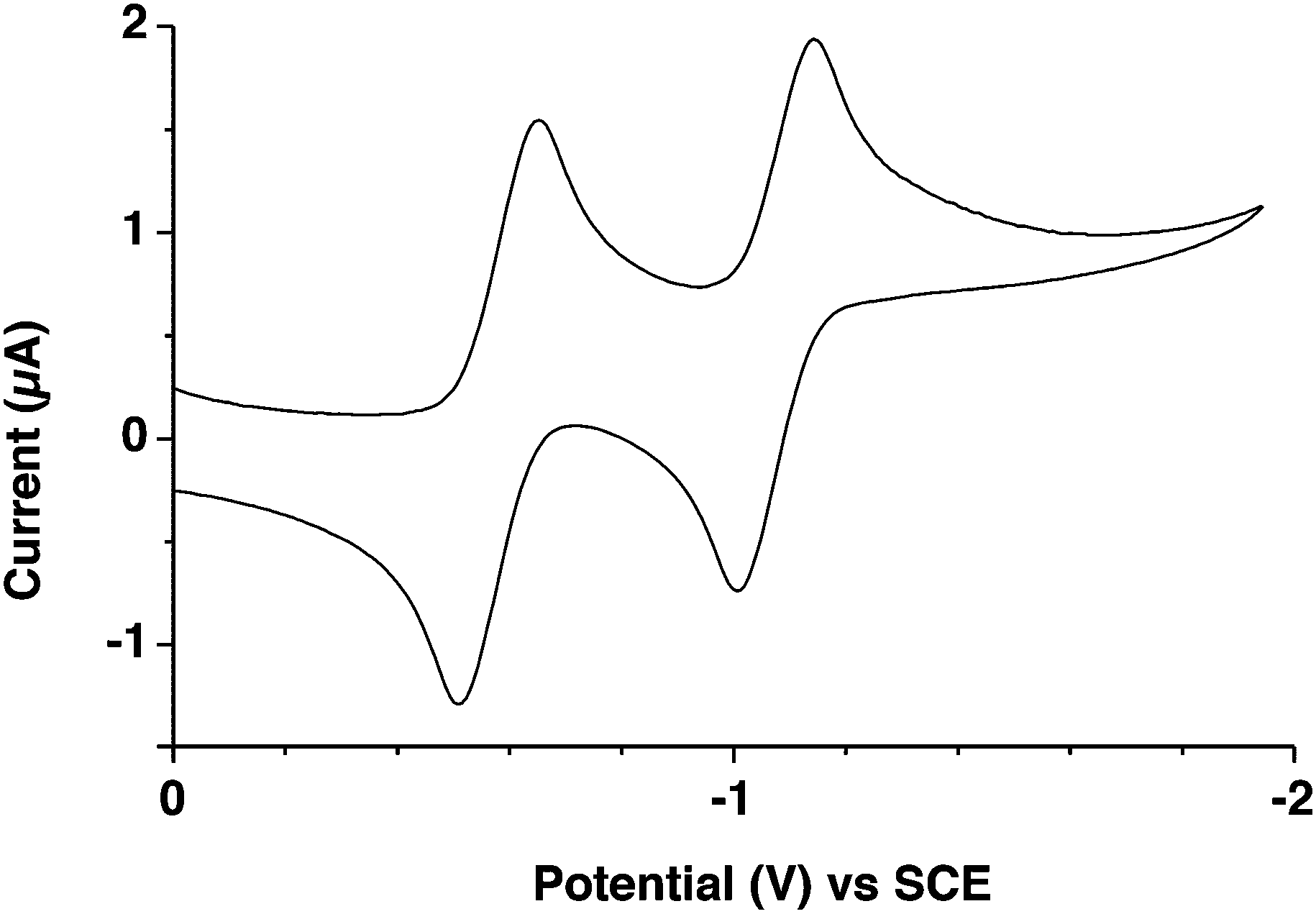 | ||
| Fig. 3 Cyclic voltammograms of pyrene-fused phenazinothiadiazole 1. | ||
The theoretical estimation (B3LYP-ODBC-6311g+(d2,p)/B3LYP-ODBC-631(d,p)) of the HOMO–LUMO gap (2.2 eV) and the LUMO (−3.78 eV) in vacuum is in good agreement with the HOMO–LUMO gap and the LUMO estimated from absorption spectroscopy and cyclic voltammetry, respectively (Table 1). Both HOMO and LUMO are delocalised along the diazonaphthothiadiazole residue, while the HOMO−1 is mostly located over the pyrene residue, which is consistent with recent reports15 (Fig. 4).
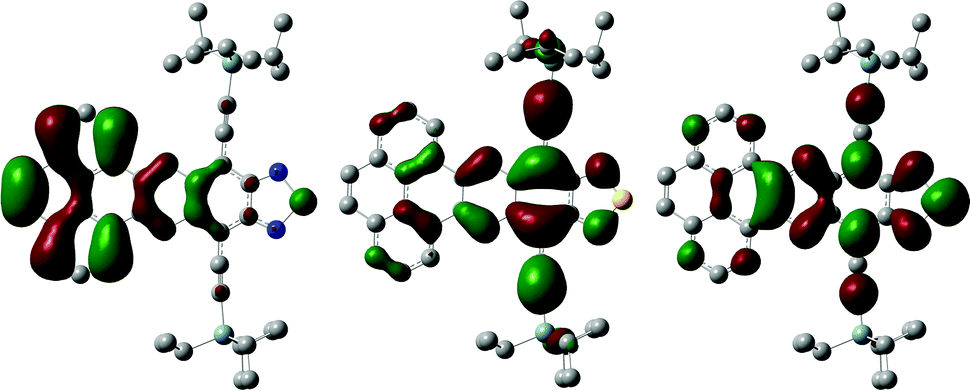 | ||
| Fig. 4 HOMO−1 (left), HOMO (center) and LUMO (right) orbitals of phenazinothiadiazole 1. C: grey, Si: cyan, H: white, N: blue, S: yellow. | ||
Thermogravimetric analysis evidences the high thermal stability of 1. No signs of decomposition were observed until 340 °C under nitrogen (Fig. S7, ESI†). In addition differential scanning calorimetry under nitrogen of 1 shows a phase transition at 121 °C that is reversible upon cooling at 117 °C (Fig. S8, ESI†).
The semiconducting properties of 1 were investigated on thin films. Bottom-contact bottom-gate transistors were fabricated on Si/SiO2 substrates on top of which gold source and drain electrodes were evaporated. Then, the SiO2 surface was treated with a layer of octadecyltrichlorosilane (OTS). Finally, 1 was vacuum-deposited at different substrate temperatures (25 °C, 80 °C and 120 °C). All the devices were characterised under vacuum after 1 h annealing at 120 °C (Table S2, ESI†). The films of 1 show a typical behaviour for n-type semiconductors (representative transfer and output curves are given in Fig. 5 and Fig. S10, ESI† respectively) with electron mobilities up to μe = 0.016 cm2 V−1 s−1 for the best performing transistor. Increasing average electron mobilities were obtained with increasing substrate temperatures during deposition (μ25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °Ce = 5.84 × 10−3 < μ80°Ce = 9.34 × 10−3 < μ120°Ce = 1.06 × 10−2).
°Ce = 5.84 × 10−3 < μ80°Ce = 9.34 × 10−3 < μ120°Ce = 1.06 × 10−2).
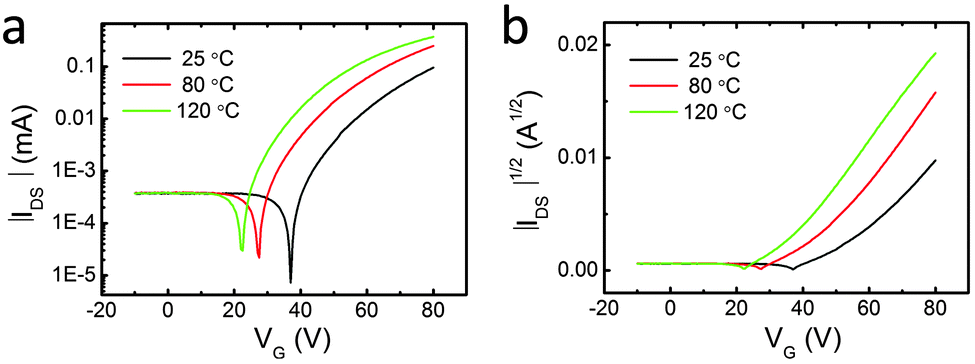 | ||
| Fig. 5 (a) Transfer curves and (b) square root of the absolute values of current as a function of gate potential for pyrene-fused phenazinothiadiazole 1 thin films deposited at different substrate temperatures. | ||
The different electron mobilities can be justified in terms of the thin film morphologies obtained upon vacuum-deposition at different substrate temperatures (Fig. 6). Pyrene-fused phenazinothiadiazole 1 spontaneously forms highly homogeneous rod-like nanostructures with aspect ratios that increase in terms of length (L25°C = ∼400 nm, L80°C = ∼860 nm and L120°C = ∼1720 nm) and diameter (∅25°C = ∼110 nm, ∅80°C = ∼110 nm, ∅120°C = ∼290 nm) with increasing substrate temperatures. After annealing at 120 °C, flake-like objects that maintain approximately the lengths of the preceding rod-like nanostructures were observed. Therefore, the larger the electron mobilities the larger is the aspect ratio of the flake-like nanostructures.
 | ||
| Fig. 6 Representative thin film morphologies (5 μm × 5 μm) of phenazinothiadiazole 1 thin films deposited at 25 °C (left), 80 °C (centre) and 120 °C (right) before (top) and after (bottom) annealing. | ||
We have described the synthesis of pyrene-fused phenazinothiadiazole 1 that exhibits a low LUMO level (−3.83 eV) and packs in slipped face-to-face 1D stacks. Thin films of 1 grown by vacuum deposition show electron mobilities (μe = 0.016 cm2 V−1 s−1) remarkably higher than those previously reported for PPA. Increasing electron mobilities were obtained with increasing substrate temperatures during deposition, which can be rationalised in terms of the larger aspect ratio of the self-assembled nanostructures obtained at higher temperatures. Overall this work demonstrates the potential of PPA to develop electron deficient materials for field-effect transistors that allow control of their properties not only at the molecular level but also at the supramolecular level.
We are grateful to the Basque Science Foundation for Science (Ikerbasque), POLYMAT, University of the Basque Country (SGIker), Deutsche Forschungsgemeinschaft (AU 373/3-1 and MA 5215/4-1), Gobierno de España (Ministerio de Economía y Competitividad, MAT2012-35826 and MAT2012-37638), Gobierno Vasco (BERC program), Diputación Foral de Guipuzcoa, the Fundação para a Ciência e a Tecnologia, ON2 (NORTE-07-0162-FEDER-000086) and the European Union (ERA-Chemistry, Career Integration Grant No. 618247, ERC Starting Grant 257654-SPINTROS and FEDER).
Notes and references
- (a) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS PubMed; (b) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed; (c) V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed; (d) H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed; (e) H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513–5517 CrossRef CAS PubMed.
- A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311–6324 RSC.
- (a) A. Mateo-Alonso, C. Ehli, K. H. Chen, D. M. Guldi and M. Prato, J. Phys. Chem. A, 2007, 111, 12669–12673 CrossRef CAS PubMed; (b) A. Mateo-Alonso, N. Kulisic, G. Valenti, M. Marcaccio, F. Paolucci and M. Prato, Chem. – Asian J., 2010, 5, 482–485 CrossRef CAS PubMed; (c) N. Kulisic, S. More and A. Mateo-Alonso, Chem. Commun., 2011, 47, 514–516 RSC; (d) S. More, R. Bhosale, S. Choudhary and A. Mateo-Alonso, Org. Lett., 2012, 14, 4170–4173 CrossRef CAS PubMed; (e) R. García, S. More, M. Melle-Franco and A. Mateo-Alonso, Org. Lett., 2014, 16, 6096–6099 CrossRef PubMed; (f) S. More, R. Bhosale and A. Mateo-Alonso, Chem. – Eur. J., 2014, 20, 10626–10631 CrossRef CAS PubMed; (g) S. More, S. Choudhary, A. Higelin, I. Krossing, M. Melle-Franco and A. Mateo-Alonso, Chem. Commun., 2014, 50, 1976–1979 RSC.
- (a) J. K. Stille and E. L. Mainen, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 665–667 CrossRef CAS PubMed; (b) J. K. Stille and E. L. Mainen, Macromolecules, 1968, 1, 36–42 CrossRef CAS; (c) K. Imai, M. Kuhihara, L. Mathias, J. Wittmann, W. B. Alston and J. K. Stille, Macromolecules, 1973, 6, 158–162 CrossRef CAS.
- (a) S. Leng, L. H. Chan, J. Jing, J. Hu, R. M. Moustafa, R. M. V. Horn, M. J. Graham, B. Sun, M. Zhu, K.-U. Jeong, B. R. Kaafarani, W. Zhang, F. W. Harris and S. Z. D. Cheng, Soft Matter, 2010, 6, 100–112 RSC; (b) J. Hu, D. Zhang, S. Jin, S. Z. D. Cheng and F. W. Harris, Chem. Mater., 2004, 16, 4912–4915 CrossRef CAS.
- (a) M. Grzelczak, N. Kulisic, M. Prato and A. Mateo-Alonso, Chem. Commun., 2010, 46, 9122–9124 RSC; (b) K. K. McGrath, K. Jang, K. A. Robins and D.-C. Lee, Chem. – Eur. J., 2009, 15, 4070–4077 CrossRef CAS PubMed; (c) K. Jang, J. M. Kinyanjui, D. W. Hatchett and D.-C. Lee, Chem. Mater., 2009, 21, 2070–2076 CrossRef CAS; (d) D. C. Lee, K. K. McGrath and K. Jang, Chem. Commun., 2008, 3636–3638 RSC; (e) D. C. Lee, K. Jang, K. K. McGrath, R. Uy, K. A. Robins and D. W. Hatchett, Chem. Mater., 2008, 20, 3688–3695 CrossRef CAS.
- (a) B. R. Kaafarani, L. A. Lucas, B. Wex and G. E. Jabbour, Tetrahedron Lett., 2007, 48, 5995–5998 CrossRef CAS PubMed; (b) J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 Search PubMed.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045–7052 CAS.
- E. Wang, L. Hou, Z. Wang, S. Hellström, W. Mammo, F. Zhang, O. Inganäs and M. R. Andersson, Org. Lett., 2010, 12, 4470–4473 CrossRef CAS PubMed.
- C. An, S. Zhou and M. Baumgarten, Cryst. Growth Des., 2015, 15, 1934–1938 CAS.
- R. C. DeCicco, A. Black, L. Li and N. S. Goroff, Eur. J. Org. Chem., 2012, 4699–4704 CrossRef CAS PubMed.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- (a) A. M. Alonso, R. Horcajada, H. J. Groombridge, R. Mandalia, M. Motevalli, J. H. P. Utley and P. B. Wyatt, Chem. Commun., 2004, 412–413 RSC; (b) A. M. Alonso, R. Horcajada, H. J. Groombridge, R. Chudasama, M. Motevalli, J. H. P. Utley and P. B. Wyatt, Org. Biomol. Chem., 2005, 3, 2832–2841 RSC; (c) A. M. Alonso, R. Horcajada, M. Motevalli, J. H. P. Utley and P. B. Wyatt, Org. Biomol. Chem., 2005, 3, 2842–2847 RSC.
- R. Garcia, M. Melle-Franco and A. Mateo-Alonso, Chem. Commun., 2015, 51, 8037–8040 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthesis, characterisation and modelling details of 1. CCDC 1056278. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc03096j |
| This journal is © The Royal Society of Chemistry 2015 |