
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the assembly of cyclotriveratrylene-derived coordination cages
James J.
Henkelis†
and
Michaele J.
Hardie
*
School of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: m.j.hardie@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6458
First published on 10th June 2015
Abstract
A review of the emerging field of cyclotriveratrylene-derived coordination cages is presented. Ligand-functionalised cyclotriveratrylene (CTV) derivatives self-assemble with a range of metal cations to afford coordination cages, polymers and topologically non-trivial constructs, such as [2]catenanes and a self-entangled cube. Increased control over their self-assembly allows for the controlled and predictable formation of well-defined coordination cages for application in host–guest and recognition chemistry, with surfactant binding and single-crystal-to-single-crystal (SCTSC) uptake of small-molecule guests being observed.
 James J. Henkelis | James J. Henkelis received his BSc, MChem and PhD degrees from the University of Leeds, UK. His doctoral research was conducted under the supervision of Professor Michaele J. Hardie on the preparation of cyclotriveratrylene derivatives, for their controlled incorporation into coordination cages and networks, towards the isolation of materials that display enhanced molecular recognition. He is currently a postdoctoral research fellow at Northwestern University (USA), working in the laboratory of Professor Sir J. Fraser Stoddart in the field of mechanostereochemistry, where his research is focussed on the synthesis and supramolecular chemistry of redox-active cyclophanes. |
 Michaele J. Hardie | Michaele J. Hardie received her BSc(Hons) and PhD degrees from the University of Melbourne, Australia with her PhD studies supervised by Dr Bernard Hoskins and Professor Richard Robson. After various postdoctoral posts including at the University of Toledo with A. Alan Pinkerton and Monash University with Colin L. Raston, she took up a lecturing position at the University of Leeds in 2001 where she is currently Professor of Supramolecular Chemistry. Her research interests are in cavitand chemistry, metallo-supramolecular chemistry, crystal engineering and small molecule crystallography. |
1 Introduction
1.1 Coordination cages
The chemistry of coordination cages has attracted a large amount of attention in recent years.1 Formed from the self-assembly of labile metal cations and suitably well-designed and pre-functionalised organic ligands, they are three-dimensional, hollow architectures that often closely resemble Platonic or Archimedean solids. In such systems, the predictability and reversibility of the coordination bond has allowed for the expedient assembly and isolation of materials that would be otherwise difficult to obtain using classical synthetic procedures. Coordination cages often possess a large and well-defined internal cavity that is chemically distinct from the bulk solution from which they are assembled. Thus, much of their chemistry has been focussed towards the binding of guest molecules or ions within these cavities and, more recently, towards their subsequent application as nanoscale reaction vessels and in enzyme-like catalysis.The capacity to perform or facilitate a chemical process within a shape-specific microenvironment requires a complementarity between the individual molecular guest components and their host, and is best realised by exacting high levels of control over the self-assembly processes. Whilst the notion of directed serendipity is a well-represented paradigm in supramolecular chemistry—and one that has been used to generate a wide range of aesthetic materials—to target a specific challenge one must be able to predictably control the self-assembly of a dynamic system with extremely high fidelity. Without control over the host there can be no control over the guest, be it a bimolecular chemical reaction, unstable reaction intermediate, or otherwise. This is a well-understood concept that extends throughout biology and the physical sciences and inspires the research in which the coherent amplification of inherent molecular properties is used to construct functional materials from simple molecular precursors.
1.2 Cyclotriveratrylene
Cyclotriveratrylene (CTV, Fig. 1) and its analogues represent a class of relatively shape-persistent C3-symmetric molecular hosts derived from the tribenzo[a,d,g]cyclononatriene scaffold.2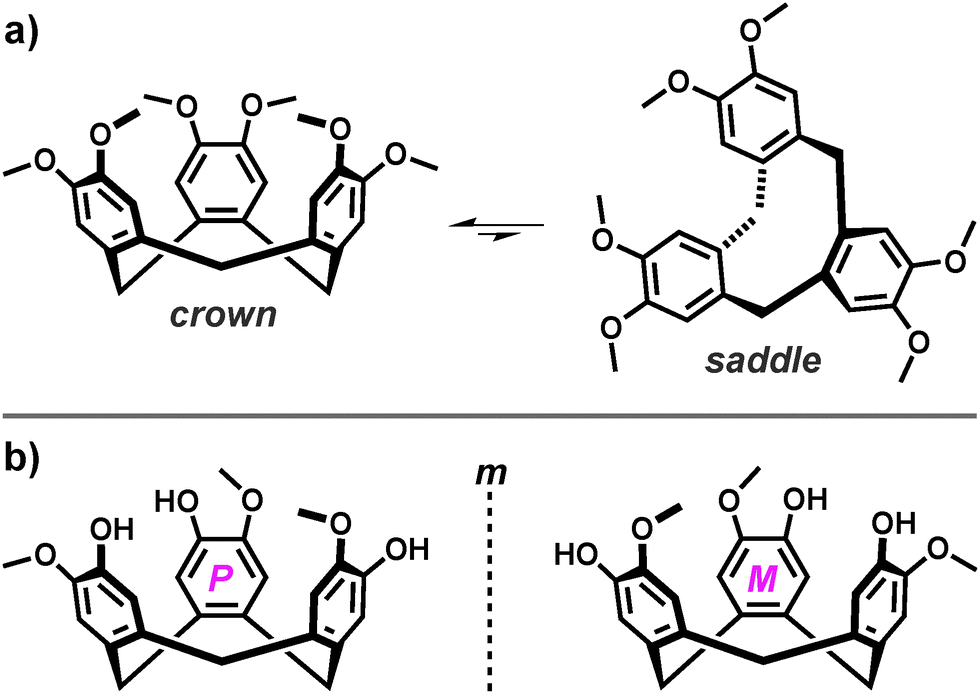 | ||
| Fig. 1 The molecular structures of (a) cyclotriveratrylene (CTV) and (b) cyclotriguaiacylene (CTG). The bowl-to-bowl inversion of CTV is indicated by the biased equilibrium between the crown and saddle molecular conformations. The two enantiomers (M and P) of CTG are shown and separated by a mirror plane (m). | ||
By virtue of their pyramidal shape, they possess an electron-rich and hydrophobic molecular cavity and exhibit interesting host–guest properties. Thus, much of their chemistry is geared towards molecular recognition. CTV forms crystalline clathrate complexes with various small organic molecules, in which the solvent guest may be contained in the molecular cavity of CTV or the CTV molecules stack in a columnar manner and the guest is contained in lattice positions.2 CTV and its analogues have also demonstrated an affinity for globular, electron-poor guests, such as fullerenes3 and carboranes,4 which form ‘ball-in-socket’ superstructures. A principal feature of CTV, and all molecular hosts,5 is an intrinsic ability to form inclusion complexes regardless of their assembly or speciation.6 With these design principles in mind, our research (and that of others) has been driven towards the controlled incorporation of molecular hosts into metallo-supramolecular constructs, in which the intrinsic hosting abilities should be transferred to the architectures afforded and, more importantly, that these properties be amplified in such a way that they can be exploited for further application, such as in catalysis and molecular separations. Whilst our work in this area has focused on analogues of CTV, other researchers have reported coordination cages derived from other types of molecular host, particularly of the calixarene family.7
Whilst supramolecular architectures of CTV itself are known,8 the vast majority of supramolecular constructs have been prepared with ligand-functionalised analogues of daughter compounds cyclotriguaiacylene (CTG), cyclotricatechylene (CTC) or cyclotriphenolene (CTP), alongside tris-amino (aCTG), tris-thiol (tCTG) and alkylated derivatives.9 Such compounds can be further derivatised through upper rim functionalisation to yield a variety of compounds, including ligands, Fig. 2.10
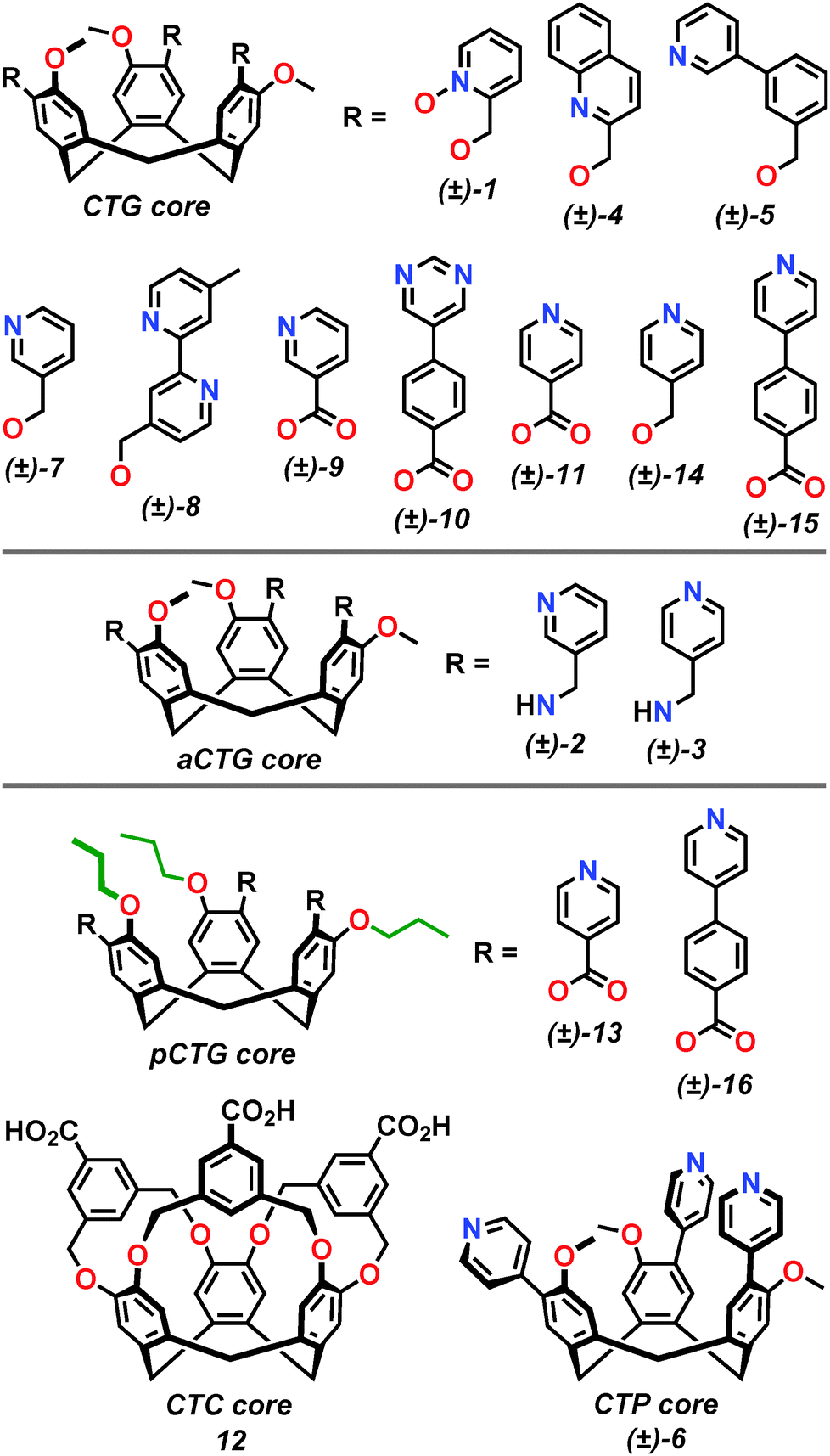 | ||
| Fig. 2 The molecular structures of ligands 1–16 referred to in this review article. Ligands are categorised according to the upper-rim functionality of the tribenzo[a,d,g]cyclononatriene core. | ||
An important feature of the tris-functionalised analogues is that they are inherently chiral and therefore exist as enantiomers.11 Whilst this becomes important upon their self-assembly, these axially chiral compounds are generally isolated, purified and used as a racemic mixture, owing to a propensity for stereochemical inversion. Although the pyramidal form of the tribenzo[a,d,g]cyclononatriene scaffold is relatively persistent, it exists in solution as a biased equilibrium of two molecular conformations that include the favoured C3-symmetric crown and the unstable saddle. The highly strained saddle conformation represents a transition state between crown-to-crown inversion, in which the three aromatic rings of the tribenzo[a,d,g]cyclononatriene scaffold are orientated orthogonally to one-another, Fig. 1a.2c,12 The rate for stereochemical inversion varies with upper-rim substitution and solvent effects, with the half-life estimated to be ca. one month (at 20 °C) based on the racemisation of deuterated analogues.13 Nevertheless, the large barrier for crown-to-crown inversion renders this process sufficiently slow at room temperature14 so that such compounds can be resolved into the corresponding enantiomers by intensive chromatographic techniques or through decoration with additional stereogenic centres.15
1.3 Cryptophanes and related organic cages
Whilst this review pertains to the construction of coordination cages, the use of CTV-type scaffolds to construct purely organic cages is a well developed field. Cryptophanes are covalently-linked capsules that are afforded through a head-to-head arrangement of two ‘CTV’ units.16 In such species, the inherent hosting ability of the tribenzo[a,d,g]cyclononatriene core is amplified by proximity and orientation effects, where guests often bind by an ‘induced fit’ mechanism.17 Generally, each ‘CTV’ fragment of a given cryptophane is axially chiral and can therefore be incorporated as one of two enantiomers, which are given the descriptors M and P, as portrayed in Fig. 3. Thus, assuming their synthesis from a racemic mixture of starting materials, the cryptophanes afforded possess two axial stereocentres, which generates four possible stereoisomers (MM, MP, PM and PP) that are comprised of two diastereoisomers, syn (MP and PM) and anti (MM and PP).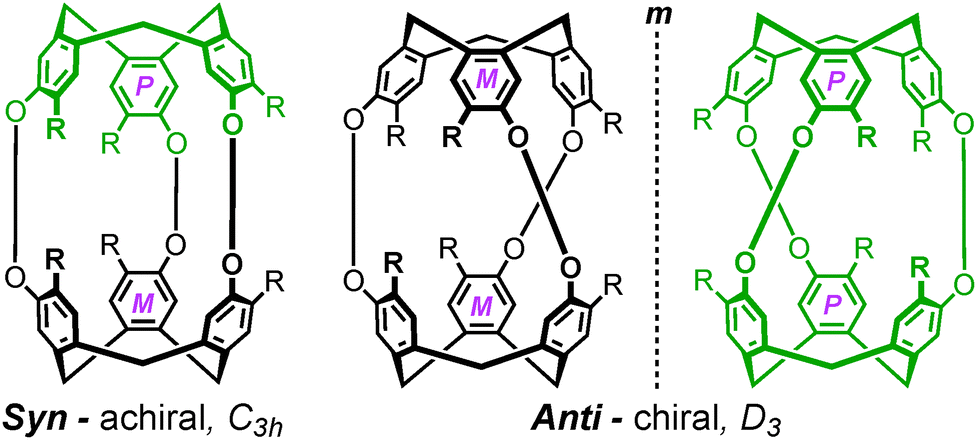 | ||
| Fig. 3 Molecular cartoons depicting the possible stereochemical configurations that result from the synthesis of traditional, organically-linked cryptophanes. Enantiomers of the tribenzo[a,d,g]cyclononatriene core are given the descriptors M and P and colour-coded for clarity. | ||
The C3h-symmetric syn diastereoisomer features the inclusion of both ‘CTV’ enantiomers and is therefore achiral as, irrespective or their relative orientation, the MP and PM stereoisomers display at least one plane of symmetry. However, the D3-symmetric anti stereoisomers MM and PP are each composed of a single ‘CTV’ enantiomer and are therefore also enantiomers. They may be described in terms of helical chirality, in which the MM and PP enantiomers possess right- (Δ) and left-handed (Λ) helicity, respectively. Of course, enantiopure cryptophanes of either Δ or Λ helical chirality can be prepared by use of enantiopure starting materials. The supramolecular stereochemistry of inherently chiral host molecules is a property that must be carefully considered in their self-assembly.18
The hosting ability of cryptophanes is proportional to their size, where the smallest bind gases, such as methane19 and xenon (with biological application in hyperpolarized 129Xe NMR),20 and the largest bind anions and small molecules, such as chloroform.21 In addition to these traditional cryptophanes, other covalently-linked constructs that include metallated analogues22 and asymmetric hemi-cryptophanes23 have been prepared. Likewise, larger species that include an octomeric nanocube have been synthesised using dynamic covalent chemistry.24
It is also worth noting that the preparation of many covalently-linked cages is challenging.25 Thus, our research focuses on the preparation of structurally analogous assemblies that are instead bridged by metallic linkers. The design principles remain the same, in that the constructs gained feature similarly well-defined and hydrophobic internal voids, although now comparatively larger, which greatly increases the scope for potential application. An attractive feature of coordination cages over their organic counterparts is that they are formed via metal-mediated self-assembly, which tends to minimize the multistep syntheses that are necessary to prepare similarly-sized organic cages and allows for their formation with relative ease. Whilst true, this statement neither accounts for serendipity, nor the fortuitous discoveries that it often affords.26
Ligand-functionalised CTV-derivatives have become increasingly well-exemplified and, as well as the coordination cages and topologically non-trivial constructs discussed in this review, a range of coordination polymers have been reported.27 This Feature Article is not a comprehensive account of the chemistry of CTV and its derivatives, and instead details the successes in the rational design and preparation of coordination cages that feature the inwardly orientated tribenzo[a,d,g]cyclononatriene scaffold for application in host–guest chemistry, in addition to a few interesting surprises that have been encountered along the way.
2 Discussion
2.1 CTV and CTC as ligands
There are no examples of coordination cages that utilise CTV itself as the ligand. The known coordination chemistry of CTV involves binding of alkali metals through the ortho-dimethoxy groups on the upper rim,8 or through the formation of organometallic π-type28 complexes—neither of which is a well-exemplified strategy for cage construction. The achiral and per-demethylated cyclotricatechylene (CTC) is more feasible as a component for building coordination cages, noting there is a rich history in the use of catechol ligands for the assembly of coordination cages,29 and early examples of metal complexes of CTC were reported by Bohle and Stasko.30Robson and coworkers have demonstrated that tetrahedral [M6L4] architectures can be constructed from CTC. Deprotonated CTC is a tris-catecholate ligand that can chelate to three transition metal centers, where the [M6(CTC–6H)4]12− anionic assemblies afforded have the CTC ligand situated at the vertices of the tetrahedra with the metal centers bridging them along each tetrahedral edge. Thus far, two noteworthy examples have been reported in cages of [Cu6(CTC–6H)4]12− and [(VO)6(CTC–6H)4]12− stoichiometry, Fig. 4.31 The copper(II) and vanadyl (V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) tetrahedra were observed in the solid state to closely associate with additional sodium and magnesium or calcium cations, respectively, and were not reported in the solution state. Similar [M6(CTC–6H)4]12− (M = Co, Mn) tetrahedra are found within complicated coordination polymers where the tetrahedral cages are linked into 3D networks by additional anion bridging that occurs between the metal centres.
O) tetrahedra were observed in the solid state to closely associate with additional sodium and magnesium or calcium cations, respectively, and were not reported in the solution state. Similar [M6(CTC–6H)4]12− (M = Co, Mn) tetrahedra are found within complicated coordination polymers where the tetrahedral cages are linked into 3D networks by additional anion bridging that occurs between the metal centres.
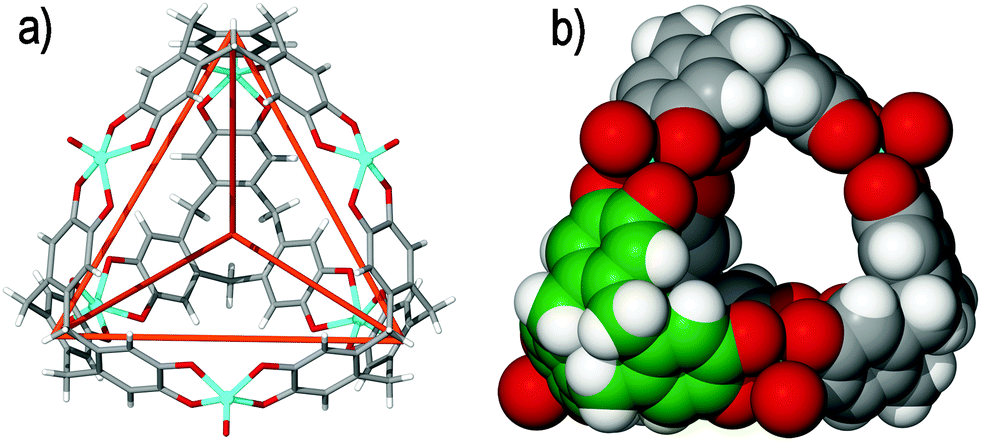 | ||
| Fig. 4 The solid state structure of Robson's [(VO)6(CTC–6H)4]12− tetrameric assembly, depicting (a) the tetrahedral cage framework, as indicated by the orange lines that connect each deprotonated CTC ligand, and (b) the space filling model, highlighting the internal void space. One CTC ligand of the assembly is colour-coded for clarity.31 | ||
Coordination cages prepared from chiral tripodal ligand-functionalised CTVs comprise the majority of known examples, which range in stoichiometry from M2L2 to M6L8 and exhibit many structural forms.32 Not all of these discrete constructs feature internal void space however, and their self-assembly is often difficult to predict and equally challenging to engineer. Thus, the following discussion represents a progression towards the controlled preparation of stable and well-defined assemblies for application in host–guest chemistry, with particular emphasis on M3L2 metallo-cryptophanes, which are directly analogous to organic cryptophanes.
2.2 M2L2 and M4L4 assemblies
Whilst M3L2 metallo-cryptophanes represent the smallest possible coordination cages that feature an accessible, internal void space, smaller constructs of M2L2 stoichiometry have been afforded. Such species are not strictly coordination cages but have been isolated in two notable forms, including an offset [Ag2(1)2]2+ capsular assembly (where 1 = (±)-tris-(2-pyridylmethyloxy-N-oxide)cyclotriguaiacylene) in which reciprocal self-inclusion is displayed, Fig. 5.33 Each ligand of the flattened assembly interacts with its partner through a ‘hand-shake’ embrace, whereby the coordinating arm of one ligand is reciprocally held in the cavity of the other through π–π and π–H interactions, and one ligand arm of each 1 ligand remains uncoordinated. Whilst this renders the capsular assembly incapable of hosting molecules, it highlights an important phenomenon that is often observed with functionalized CTVs, which is their propensity to recognize one another in the solid state.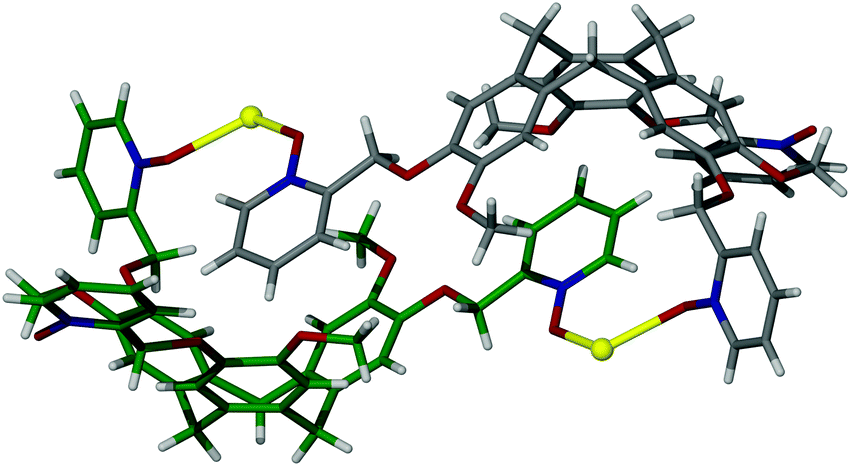 | ||
| Fig. 5 The skeletal structure of complex [Ag2(1)2]2+, evidencing the flattened ‘handshake’ motif. One of the two 1 ligands is colour-coded and all solvent molecules and anions are omitted for clarity.33 | ||
Whilst this is testament to their hosting ability, it demonstrates just how easily their self-assembly can be perturbed and how challenging their self-assembly can be to predict. Similar offset M2L2 assemblies of derivatised CTVs have been observed in both coordination34 and organometallic35 complexes, alongside purely covalent species.36
The second noteworthy complex of M2L2 stoichiometry is the dimeric [Ag2(2)2(MeCN)2]2+ capsule (where 2 = (±)-tris-(3-pyridylmethylamino)cyclotriguaiacylene), which is formed from the self-assembly of ligand 2 and silver(I) hexafluorophosphate (PF6−) in acetonitrile (MeCN) solvent, Fig. 6a.37
 | ||
| Fig. 6 The solid state structures of (a) the dimeric capsular assembly [Ag2(2)2(MeCN)2]2+ and (b) the [Ag4(3)4(MeCN)4]4+ tetrahedral cage. Non-covalently bound MeCN guests are coloured orange and displayed in space-filling mode in each case for clarity. The tetrahedral framework of [Ag4(3)4(MeCN)4]4+ is denoted by the use of solid, yellow lines that connect the base of each 3 ligand.37 | ||
The [Ag2(2)2(MeCN)2]2+ capsule more closely resembles a metallo-cryptophane than the aforementioned flattened [Ag2(1)2]2+ assembly owing to the head-to-head arrangement of ligands. The two silver(I) centres that bridge the complex are tetrahedrally coordinated by both 2 and acetonitrile ligands, in which the latter coordinate endo to the cage such that the terminal methyl group of the coordinating acetonitrile molecule forms host–guest interactions with the tribenzo[a,d,g]cyclononatriene core of each ligand. Hence, solvent host–guest templation is an important factor in complex formation.
Solvent host–guest interactions38 were also identified when using the positional isomer (±)-tris-(4-pyridylmethylamino)cyclotriguaiacylene 3 under analogous self-assembly conditions, in which an expanded [Ag4(3)4(MeCN)4]4+ tetrahedral assembly was afforded, Fig. 6b.37 Tetrahedral coordination cages are well exemplified in the literature and their self-assembly, physicochemical properties and hosting phenomena have been intensively studied.29,39 Each pyramidal ligand of [Ag4(3)4(MeCN)4]4+ represents one of the tetrahedral vertices that coordinate the four trigonal silver(I) centres at each of the tetrahedron's faces. The tetrahedral cage plays host to additional uncoordinated solvent molecules that interact with each of the exposed ligand cores in a manner similar to that described for the smaller [Ag2(2)2]2+ capsule. Each [Ag4(3)4(MeCN)4]4+ tetrahedral assembly is formed from a single ligand enantiomer, in spite of the racemic mixture used for self-assembly, and is therefore homochiral. However, the bulk mixture is that of a racemate, with an equal proportion of enantiopure cages composed of the M and P ligand enantiomers. The cage was observed to exist both in solution and in the crystalline solid state.
Many tetrahedral coordination cages display a propensity to bind anionic or cationic guests and the hosting ability of the [Ag4(3)4(MeCN)4]4+ tetrahedral assembly towards anions was investigated, but inclusion complexes were not observed. Carborane anions were included in the investigation; however, only exo-association was identified and the molecular cavity remained occupied with the acetonitrile guest.40 Interestingly, if the larger glutaronitrile is added to the reaction mixture as a potential guest, the self-assembly product is a 2D coordination polymer over the free cage, in which the glutaronitrile associates with the molecular cavity. As described above, self-assembly of AgPF6 and ligand 2 gives the pinched [Ag2(2)2(MeCN)2]2+ capsule, but use of AgSbF6 and an acetonitrile–acetone solvent mixture gives rise to a coordination polymer with no inclusion of acetonitrile. Thus, capsule formation is only observed in these [AgnLm]n+ systems in the presence of host–guest interactions to acetonitrile.41
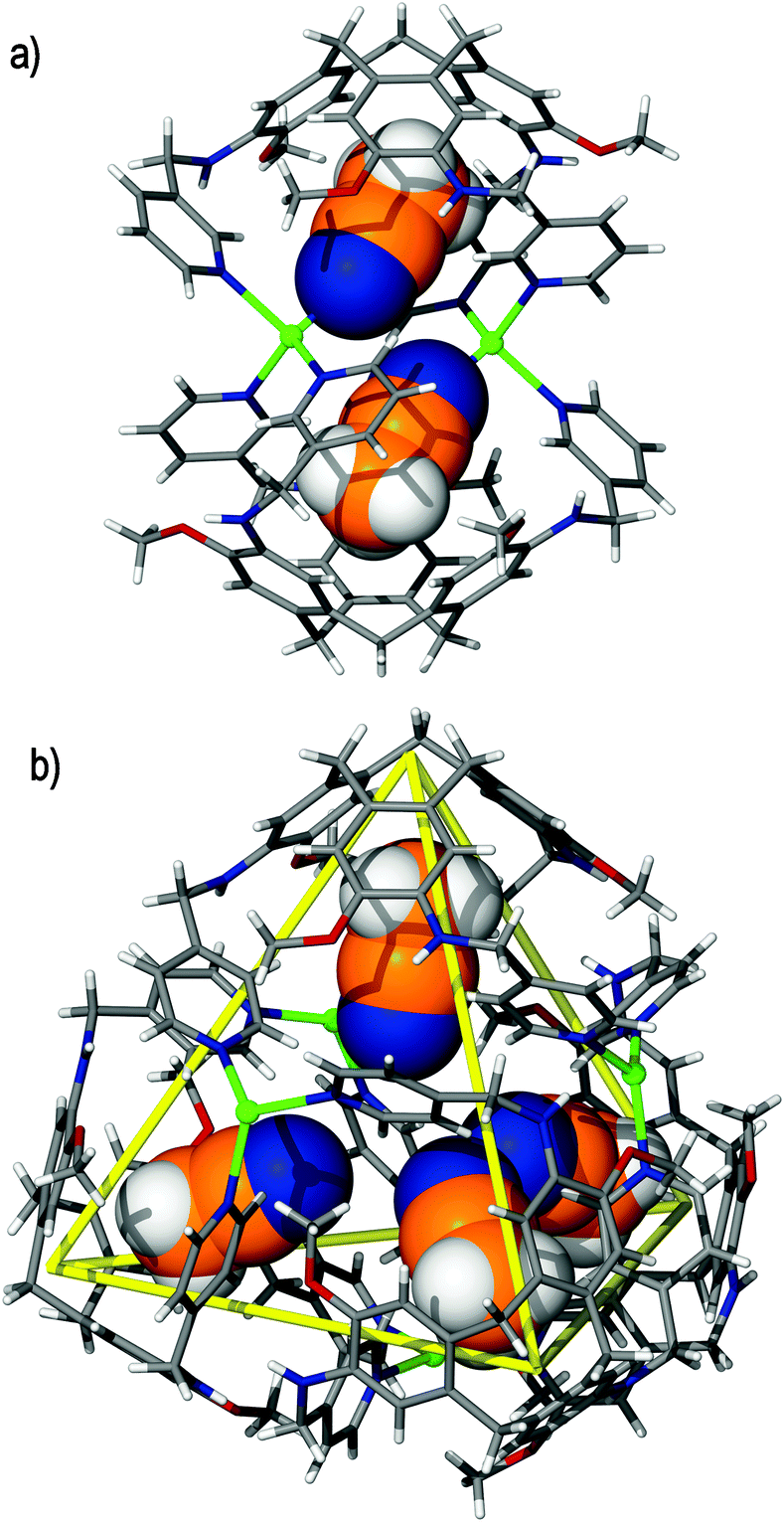
Other constructs of M4L4 stoichiometry have also been afforded in a [Ag4(4)4]4+ twisted tetrahedron42 and [Pd4(5)4]8+ Solomons cube.43 The [Ag4(4)4]4+ cage (where 4 = (±)-tris-(2-quinolylmethyloxy)cyclotriguaiacylene) forms only in the solid state and takes the form of a tetrameric cuboid in which four 4 ligands coordinate to four independent silver(I) cations in a tetrahedral array, Fig. 7. In this example, the silver(I) centres are linearly coordinated by the four ligands, which coordinate through only two of their quinolyl donors in a twisted manner and lie along each the face of the tetrahedron. The [Ag4(4)4]4+ assembly is achiral and exists as a dimer-of-dimers, in which there are two M and two P ligand enantiomers present in the resultant cage.
 | ||
| Fig. 7 The solid state structure of the tetrameric [Ag4(4)4]4+ twisted tetrahedron, depicting (a) the tetrahedral arrangement of silver(I) cations by use of solid, yellow lines and (b) the resultant cubic structure, as viewed in space-filling mode. One 4 ligand in each view of the assembly is coloured pink for clarity.42 | ||
Whilst complex [Ag4(3)4(MeCN)4]4+ is afforded through acetonitrile solvent templation, complex [Ag4(4)4]4+ assembles in 2,2,2-trifluoroethanol (TFE) solvent and shows no solvent guest binding. Rather, its formation is facilitated by reciprocal self-inclusion, in which the relatively large aromatic surface of each quinolyl ligand arm plays guest to the molecular cavity of a proximal ligand, giving the overall assembly a negligible internal void space.
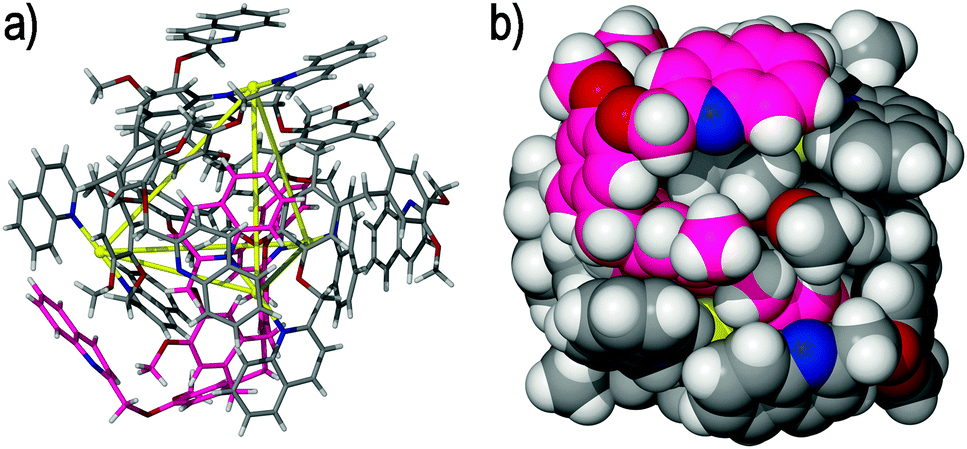
Although not a classical coordination cage, the [Pd4(5)4(NO3)2(H2O)2]6+ Solomons cube (where 5 = (±)-tris-(3-(3-pyridyl)benzoyl)cyclotriguaiacylene) is a unique and interesting assembly.43 The complex was prepared from the self-assembly of ligand 5 and palladium(II) nitrate (NO3−) in dimethylsulfoxide (DMSO) solution. Here, the interweaving and twisting of ligands affords a self-entangled structure that contains elements of a Solomon's link, including two ring motifs that feature alternating over and under crossing points within the structure, Fig. 8. The four palladium(II) centres represent the corners of a square that are connected by four ligands. Tracing the connectivity between the molecular components highlights four topological crossing points that render the [Pd4(5)4(NO3)2(H2O)2]6+ tetramer topologically non-trivial.
 | ||
| Fig. 8 The topologically complex [Pd4(5)4(NO3)2(H2O)2]6+ Solomons cube, highlighting (a) the molecular structure and four topological crossing points within the assembly (shown as yellow lines bridging the palladium(II) nodes), and (b) the two figure-of-eight motifs found within the assembly, which are colour-coded and simplified for clarity.43 | ||
What is most interesting is its spontaneous formation by self-assembly processes, unlike the template-directed procedures most often employed to form topologically nontrivial assemblies.44 The Solomon's cube represents the most complex architecture yet identified with derivatised CTVs and packs in the solid state to afford a hollow spheroid that closely resembles an Archimedean truncated hexahedron. The complex was observed to exist both in solution and the crystalline solid state. An interesting feature of the [Pd4(5)4(NO3)2(H2O)2]6+ Solomons cube is its homochirality, where each interlocked component possesses the same absolute stereochemistry.
Whilst architecturally aesthetic, the above examples further illustrate the unpredictable nature of the self-assembly of ligand-functionalised CTVs and highlight the necessity for increasingly rigorous control over their self-assembly processes, if shape-specific cages are to be obtained.
2.3 Metallo-cryptophanes: expected and unexpected behavior
In 2001 Shinkai, Yamaguchi and co-workers reported the first CTV-based coordination cage, comprising a head-to-head arrangement of tris(4-pyridyl)CTG 6 ligands, linked through metal coordination by three cis-protected palladium(II) metal centres.45 Use of a mixture of ligand enantiomers affords a mixture of cage stereoisomers, namely both enantiomers of the chiral anti-isomer as well as the meso syn-isomer, which are observed to interconvert in solution. The anti-[Pd3(6)2]6+ metallo-cryptophanes can be selectively prepared by using the enantiopure ligand, Fig. 9. | ||
| Fig. 9 Cartoon depictions of the two enantiomers of Shinkai's anti-[Pd3(6)2]6+ metallo-cryptophane, as separated by a mirror plane (m).45 | ||
These cages feature a well-defined internal void space that is distinct from the bulk solution from which they are formed, but their hosting capabilities were not explored. These metallo-cryptophanes first indicated the amenability of ligand-functionalised CTVs for the preparation of coordination cages that may exhibit inclusion phenomena as a result of the inwardly orientated tribenzo[a,d,g]cyclononatriene ligand scaffold. Trigonal bipyramidal coordination cages of M3L2 stoichiometry have been reported with other classes of tripodal ligand but are not widespread.46
Taking inspiration from Yamaguchi and Shinkai's earlier research,45 one could envisage two possible routes towards a M3L2 metallo-cryptophane, in which ligand-functionalised CTVs are decorated with either 4- or 3-pyridyl (or equivalent) donor moieties to be linked by 90 and 180° metallic tectons, respectively. We initially investigated the alternative design to that exemplified by Shinkai and Yamaguchi's by pursuing the cages prepared from the self-assembly of 3-pyridyl-decorated CTVs with a 180° linker.
It was predicted that ligand (±)-tris-(3-pyridylmethyloxy)cyclotriguaiacylene 7 would afford a metallo-cryptophane upon self-assembly with a suitably linear metal centre. The self-assembly of ligand 7 and silver(I) perchlorate (ClO4−) in N,N′-dimethylformamide (DMF) solution does indeed afford a metallo-cryptophane, although not the one that was anticipated. Rather, a topologically complex and triply-interlocked [2]catenane was isolated, of [Ag6(7)4]6+ stoichiometry, comprising two mechanically-interlocked, but chemically independent, anti-[Ag3(7)2]3+ metallo-cryptophanes, Fig. 10.47
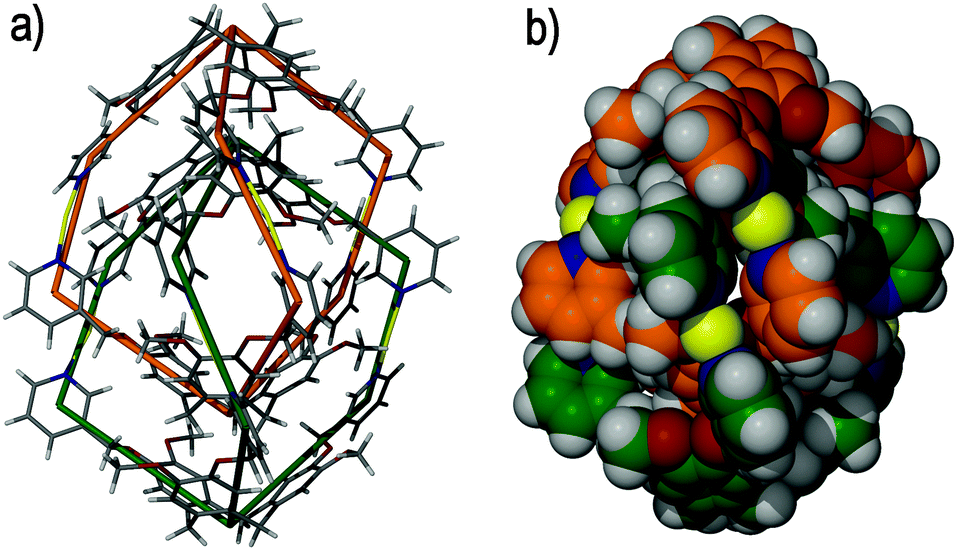 | ||
| Fig. 10 The solid state structure of the triply-interlocked [2]catenane [Ag6(7)4]6+, in which only the (Λ,Λ)-[Ag6(7)4]6+ enantiomer is shown. The mechanically interlocked components are shown as (a) wire frames and (b) in space filling mode. In both illustrations the two chemically independent metallo-cryptophanes are colour-coded for clarity.47 | ||
Each trigonal bipyramidal anti-[Ag3(7)2]3+ cage is composed of two 7 ligands and is bridged, as expected, by approximately linear silver(I) cations with the ligands in a head-to-head orientation. The inclusion of a single ligand enantiomer affords each chiral metallo-cryptophane, with the P ligand enantiomer giving the left-handed helical (Λ)-[Ag3(7)2]3+, and the M ligand enantiomer giving the right-handed helical (Δ)-[Ag3(7)2]3+.
The individual [Ag3(7)2]3+ cryptophanes interlock in a chiral manner to afford an equal mixture of (Δ,Δ)-[Ag6(7)4]6+ and (Λ,Λ)-[Ag6(7)4]6+ enantiomers. The (Δ,Δ)-[Ag6(7)4]6+ and (Λ,Λ)-[Ag6(7)4]6+ enantiomers differ only in their absolute stereochemical configuration. These form in the absence of a template, and there are no formal interactions between each metallo-cryptophane of the [2]catenane. This is relatively unusual, as template-directed procedures are often required to reliably install a mechanical bond within such metallo-supramolecular complexes.48 Catenation in this instance is likely entropically driven,49 where the interlocking of each [Ag3(7)2]3+ cage expels high energy solvent from within the cavity, although symmetry-driven self-recognition may also play a role.50 It is remarkable that the resultant (Λ,Λ)-[Ag6(7)4]6+ and (Δ,Δ)-[Ag6(7)4]6+ [2]catenanes are homochiral and that the overall helical chirality and D3-symmetry are conserved, especially given their formation from a racemic mixture of ligands and in the absence of a template. The selective formation of homochiral complexes from a racemic mixture of molecular components is particularly noteworthy.51 Alterations to the reaction conditions did not change the self-assembly processes and spontaneous catenation was always observed. Nevertheless, the serendipitous formation of the [2]catenane suggests a complex threading and interlocking mechanism that is still not very well understood.
The internal volume of the [2]catenane was estimated from its crystal structure and calculated to be 197 Å3. Regardless, there are no solvent molecules located within the cavity and there are limited windows to allow for guest exchange. Interpenetration of the individual [Ag3(7)2]3+ metallo-cryptophanes significantly reduces the internal space available to host molecules and, by considering the packing requirements according to Rebek and co-workers,52 estimates an ideal guest size of 108 Å3, which renders the cage cavity too small for further host–guest application other than for small gases.
To date, only one other example of catenation has been identified with ligand-functionalized CTVs, which results from the spontaneous interlocking of (±)-tris-(4-(4′-methyl-2,2′-bipyridyl)-benzyloxy)cyclotriguaiacylene 8 and zinc(II) nitrate in DMSO solution. The resultant (±)-[Zn6(8)4(NO3)6]6+ [2]catenane features a similar 3D appearance that is comprised of two interlocking syn-[Zn3(8)2(NO3)3]3+ metallo-cryptophanes, Fig. 11.53 In this example each syn-[Zn3(8)2(NO3)3]3+ metallo-cryptophane is inherently chiral in spite of the inclusion of both ligand enantiomers owing to the left-handed helical chirality (Λ) about the zinc(II) coordination environment, which removes the horizontal mirror plane expected of a syn-diastereoisomer.54 The resultant [2]catenane therefore displays planar chirality, owing to the inequivalence of the PMMP and MPPM stereoisomers that are afforded through interlocking of the individual syn-[Zn3(8)2(NO3)3]3+ metallo-cryptophanes. Unlike the anti-[Ag6(7)4]6+ [2]catenane discussed above, the (±)-[Zn6(8)4(NO3)6]6+ [2]catenane features six instances of weak hydrogen bonding between the mechanically-interlocked components, which may imply that some degree of self-templation facilitates its formation.55 The first report of a cage catenane species was from Fujita and co-workers,56 followed a decade later by our report of (±)-[Zn6(8)4(NO3)6]6+ and its Co(II) analogue. Since then there have been a small flurry of reports of coordination cage catenanes57 and their organic counterparts.58
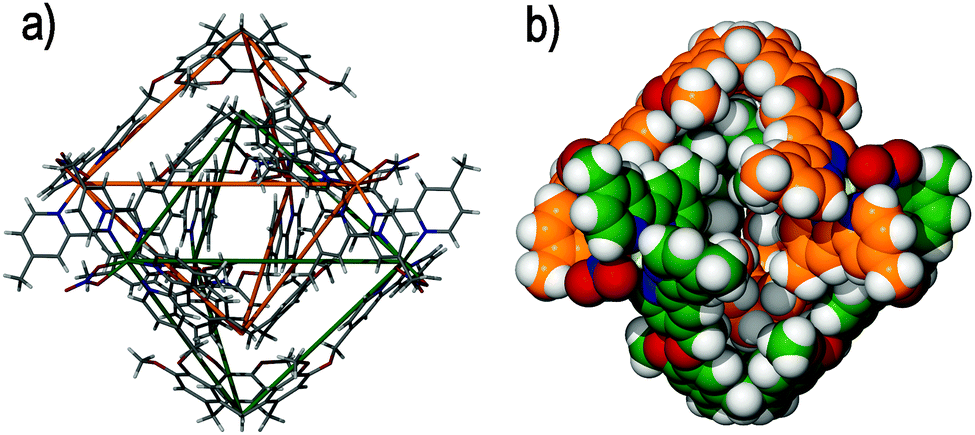 | ||
| Fig. 11 Taken from the crystal structure of the triply-interlocked [2]catenane (±)-[Zn6(8)4(NO3)6]6+, in which the two mechanically interlocked metallo-cryptophanes are (a) displayed in skeletal form with the topological crossing points denoted by the green and orange wire frames, and (b) colour-coded and displayed in space filling mode.54 | ||
The flexibility of ligand 7 was assumed to be a contributing factor to the catenation described above. It was therefore predicted that ligand (±)-tris-(3-carboxypyridyl)cyclotriguaiacylene 9, which features the same 3-pyridyl functionality but linked through a rigid ester linkage, would afford the desired metallo-cryptophane, sans-catenation. Irrespective of the conditions employed, all attempts to form the metallo-cryptophane were unsuccessful, perhaps as a result of poor ligand solubility. The self-assembly of the extended ligand (±)-tris(4-(5-pyrimidyl)benzoyl)cyclotriguaiacylene 10 under the same reaction conditions afforded crystals of the chiral anti-[Ag3(10)2]3+ metallo-cryptophane, in which two 10 ligands are orientated in the desired head-to-head manner and bridged by three linear silver(I) centres, Fig. 12a.47 Unfortunately, whilst the anti-[Ag3(10)2]3+ cage possesses a large and accessible internal void space, the weak Lewis basicity of the pyrimidine donor prevents its solution-phase existence and renders it a phenomenon of the solid state. Moreover, the resultant cage windows are too large to afford a suitably distinct microenvironment, which would likely impede guest encapsulation. The evident inability of ligands 7–10 to self-assemble with a suitably linear metal centre to afford the corresponding single-cage M3L2 metallo-cryptophane was mirrored in the self-assembly of positional isomer (±)-tris-(4-carboxypyridyl)cyclotriguaiacylene 11 with cis-protected palladium(II) salts. Despite Yamaguchi's literature precedent45 their self-assembly did not furnish the desired capsular assembly, which again may be due to the relatively low solubility level of the ligand.
 | ||
| Fig. 12 The solid state structures of (a) the chiral anti-[Ag3(10)2]3+ metallo-cryptophane,47 (b) the ‘bow-tie’ metallo-cryptophane, [{Cu3(12)2(DMF)(H2O)}2(BPE)],59 and (c) tribenzotriquinacene triacid based [Cd3L2] metallo-cryptophane.63 | ||
Metallo-cryptophane formation is not necessarily limited to pyridyl donors, and a ‘bow-tie’ analogue has been afforded through the self-assembly of ligand tris-(3,5-bis-(methyl)benzoic acid)cyclotricatechylene 12 and basic metal salts, including copper(II) and cobalt(II) acetate.59 Here, in situ deprotonation of the tris-carboxylate ligand is afforded solvothermally in the presence of either DMF or DEF (N,N′-diethylformamide) solvents. The constructs afforded are not strictly metallo-cryptophanes, but do feature the required head-to-head arrangement of 12 ligands. Complex [Cu3(12)2(DEF)2] is charge neutral and has a pinched appearance, owing to the bridging metallic cluster, and therefore features no significant accessible void space. Likewise, complex [Co7(μ3-12)2(μ4-O)2(DMF)2] is again pinched at the centre, yet may display potentially magnetic properties owing to the unusual Co7 core.60 The interesting feature of these multinuclear complexes is the presence of labile solvent ligands about the bridging metallic cores. For example, the DEF ligands in complex [Cu3(12)2(DEF)2] are easily displaced by ligand bis(4-pyridyl)ethylene (BPE) to afford a dimer of M3L2 metallo-cryptophanes and furnish the named ‘bow-tie’ motif, of [{Cu3(12)2(DMF)(H2O)}2(BPE)] stoichiometry, Fig. 12b. Carboxylate-functionalised CTVs are not as well exemplified as their N-donor counterparts, yet have shown the propensity for Cu(II) in a tubular 1D coordination polymer reported by Zheng and coworkers,61 and for lanthanide(III) coordination in the formation of a europium(III)-containing 2D network with a decorated Kagome Dual (kgd) topology.62 Cao, Kuck and co-workers have recently reported a [Cd3L2] metallo-cryptophane where L is a C3v-symmetric tris-carboxylate decorated tribenzotriquinacene cavitand ligand, Fig. 12c.63 The [Cd3L2] metallo-cryptophane has a similar pinched-in aspect to [Cu3(12)2(DEF)2].
Whilst the complexes outlined above can be classed as metallo-cryptophanes, only the prototype reported by Shinkai and Yamaguchi represents a promising candidate for extensive host–guest applications. The unpredictability of their self-assembly is again evidenced in the serendipitous isolation of [2]catenane species, and in the isolation of complexes that are only accessible in the solid state, or those which do not possess meaningful voids as a result of bridging metal clusters. Nevertheless, their isolation affords valuable insight as to what may be necessary for their predictable formation and isolation.
2.4 The shape-shifting metallo-cryptophane
The unsuccessful self-assembly of ligand 11 with cis-protected palladium(II) salts identified that a fine balance of rigidity and solubility were necessary to facilitate the formation of a M3L2 metallo-cryptophane. To address this, pCTG (a propylated analogue of CTG) was used to prepare ligand (±)-tris-(4-carboxylpyridyl)-tris-(propoxy)cyclotricatechylene 13, which afforded significantly improved ligand solubility in a wide range of solvents.64Whilst methylated ligand 11 was not observed to form a stable metallo-cryptophane with cis-protected palladium(II) salts, the reaction of its propylated congener 13 with [Pd(en)(NO3)2] (en = ethylenediamine), in DMSO solvent, afforded the desired [Pd3(en)3(13)2]6+ metallo-cryptophane. Unfortunately, the resultant complex was observed to be a metastable product of self-assembly. It undergoes a significant structural and compositional reorganisation from the [Pd3(en)3(13)2]6+ metallo-cryptophane to a [Pd6(13)8]12+ octahedral coordination cage, which is assumed to be the thermodynamic product.7 It is worth noting that intracage transformations such as these remain relatively rare;65 however, a near identical transformation was previously reported by Chand and co-workers. They reported that ligand-exchange occurred for the trigonal prismatic [Pd3(en)3(tpb)2]6+ cage, where tpb = 1,3,5-tris(4-pyridylmethyl)benzene, which transforms to the sphere-like [Pd6(tpb)8]12+ assembly.66 The ethylenediamine auxiliary ligand has been well exemplified as a cis-protecting ligand and used as a design feature to control available ligation sites at the metal centre in a wide range of metallo-supramolecular assemblies.67 Thus, its displacement in these examples implies that the driving force for cage expansion exceeds the kinetic barrier for its dissociation from the palladium(II) centre.68
Although this inter-cage transformation was unexpected, the resultant [Pd6(13)8]12+ assembly was easy to identify as we had previously observed its methylated congener from the self-assembly of ligand 11 and palladium(II) salts in DMSO solution as the [Pd6(11)8]12+ ‘stella octangula’ cage, which will be discussed in more detail in section 2.6.69 In spite of their inherent similarity, it remains unclear why ligand 13 would afford a metastable [Pd3(en)3(13)2]6+ metallo-cryptophane en route to the [Pd6(13)8]12+ cage whereas ligand 11 would not.
2.5 Metallo-tecton-directed assembly of metallo-cryptophanes
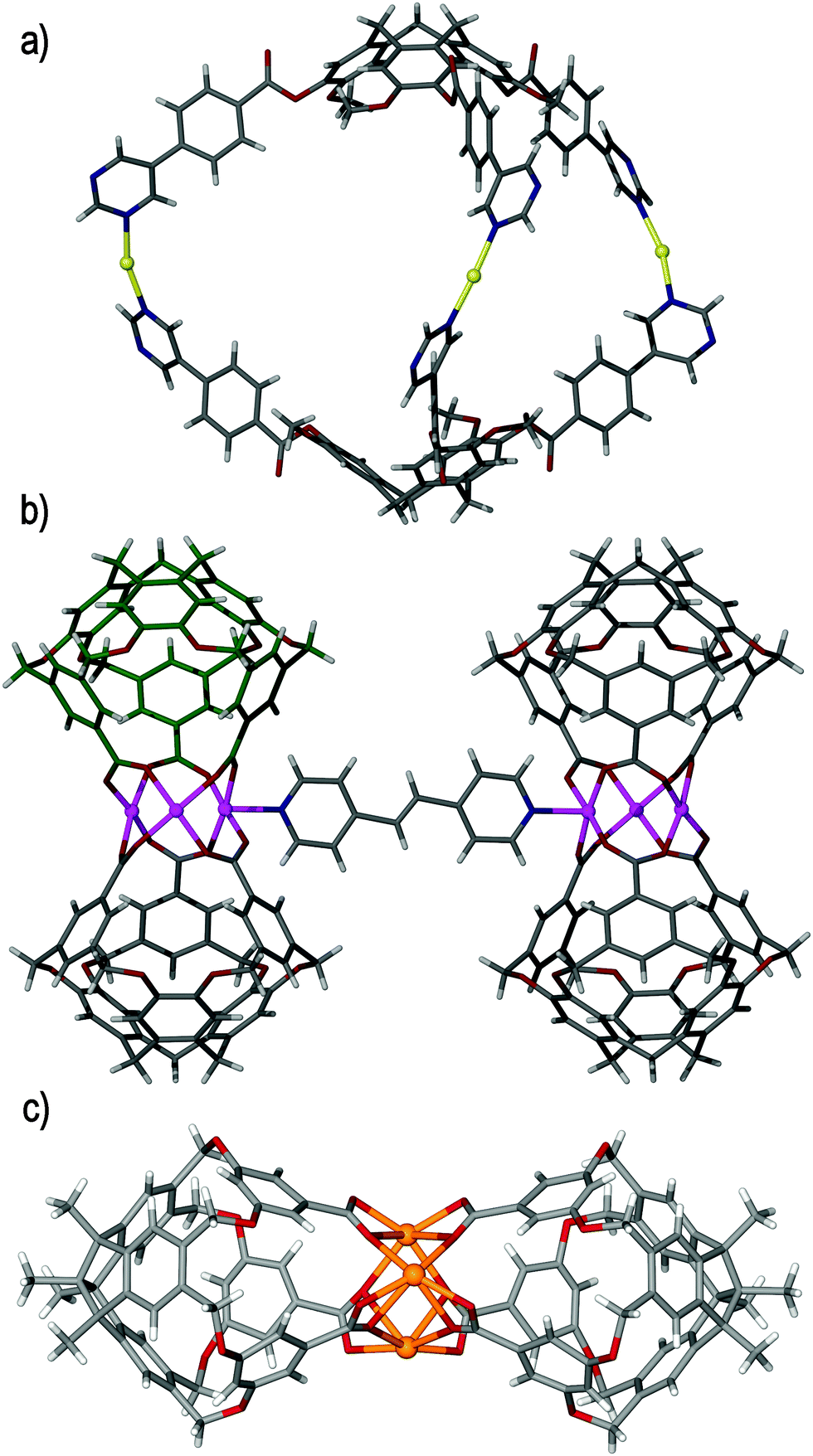
The existence of the metastable [Pd3(en)3(13)2]6+ metallo-cryptophane highlighted that a stable M3L2 metallo-cryptophane may be reliably assembled with ligands such as 13 if an appropriate cis-protected palladium(II) salt can be found. The self-assembly of ligand 13 was investigated with a wide range of cis-protected palladium(II) salts, with both diphosphine (as employed by Shinkai and Yamaguchi) and N-donor auxiliary ligands proving incapable of facilitating the formation of the metallo-cryptophane. To address this, a bis-NHC–palladium(II) tecton (where NHC = N-heterocyclic carbene) decorated with naphthylene moieties was utilised. Although metal complexes of NHCs are more commonly associated with catalysis70 than with supramolecular self-assembly, they offer the necessary kinetic stability required to prevent dissociation from the metal centre. Moreover, such complexes promote trans-labilisation at the metal centre which facilitates self-assembly and helps drive the equilibrium towards a thermodynamic minimum.71 Some examples metallo-supramolecular constructs are known.72The self-assembly of ligand 13 and the metallo-tecton, [Pd(Nap)(NCMe)2]2+ (where Nap is a 2-naphthylene derived bis-N-heterocyclic carbene) in nitromethane solvent rapidly and quantitatively afforded the desired [Pd3(Nap)3(13)2]6+ metallo-cryptophane,73 which was observed as the only product of self-assembly and did not undergo further equilibrium or structural reorganisation.7 Corroboration of the solution-phase and solid state structures of the [Pd3(Nap)3(13)2]6+ metallo-cryptophane evidenced the formation of a single diastereoisomer (syn), which featured the inclusion of both M and P ligand enantiomers, Fig. 13a. The controlled assembly and enhanced stability of the syn-[Pd3(Nap)3(13)2]6+ metallo-cryptophane was attributed to the extensive aromatic interactions afforded between the cage and naphthyl tecton, which were evidenced to lock the complex in place, once formed. These interactions were shown to be highly persistent, with the syn-[Pd3(Nap)3(13)2]6+ metallo-cryptophane remaining intact upon heating to 80 °C in nitromethane solution.
 | ||
| Fig. 13 Taken from the crystal structure of (a) the parent syn-[Pd3(Nap)3(13)2]·6(BF4) metallo-cryptophane, displayed in space-filling mode and with each 13 ligand of the head-to-head assembly distinguishable by colour and the cis-protecting NHC auxiliary coloured green for clarity. Host–guest inclusion complexes are displayed in (b) syn-[Pd3(Nap)3(13)2]·6(BF4)·3(DCB) and (c) syn-[Pd3(Nap)3(11)2]·6(BF4)·3(I2), in which the metallo-cryptophanes are shown in skeletal form and the DCB and iodine guests (some disordered positions excluded)are shown in space-filling mode and coloured pink and purple, respectively.73 | ||
The syn-[Pd3(Nap)3(13)2]6+ metallo-cryptophane features a head-to-head arrangement of ligands, in which the inwardly orientated tribenzo[a,d,g]cyclononatriene ligand scaffolds afford the shape-persistent and hydrophobic internal cavity necessary for subsequent host–guest studies, with inclusion phenomena being exhibited in the crystalline solid state. The solid state structure of the syn-[Pd3(Nap)3(13)2]·6(BF4) metallo-cryptophane indicated that individual cages interact through a network of C–H⋯F hydrogen bonds between the syn-[Pd3(Nap)3(13)2]6+ assembly and BF4− (tetrafluoroborate) anions. This acts to propagate the [Pd3(Nap)3(13)2]6+ metallo-cryptophanes in two dimensions, resulting in a network of cages in which the molecular cavities represent periodic and well-defined voids within the crystal lattice, although there were no significant channels between the cages. Isostructural crystalline metallo-cryptophane complexes were also isolated from ligand 11, along with other CTV-type ligands, which were equally robust towards changes in temperature and pressure and could be evacuated by heating under vacuum.
Single crystals of the syn-[Pd3(Nap)3(13)2]·6(BF4) metallo-cryptophane were observed to be mechanically robust and stable towards desolvation. Whilst classically non-porous, they were observed to uptake small molecules in a single-crystal-to-single-crystal (SCTSC) fashion. Submersion of the crystalline syn-[Pd3(Nap)3(13)2]·6(BF4) networked metallo-cryptophane in a solution of 1,2-dichlorobenzene (DCB) saw the crystals darken in colour as the guest was absorbed. Single crystal diffraction analysis evidenced guest uptake, in which the DCB guests were located exo to the metallo-cryptophane and bound in the smaller, interstitial sites between cage windows, Fig. 13b. It was believed that the crystal lattice permutes sufficiently to allow for the diffusion of guest through the network, driven by van der Waals cooperativity.6a The extent of DCB uptake was evidenced by 1H NMR spectroscopy, thermogravimetric analysis (TGA) and energy-dispersive X-ray (EDX) analysis, where the complex stoichiometry was determined to be syn-[Pd3(Nap)3(13)2]·6(BF4)·3(DCB).
Submerging single crystals of the syn-[Pd3(Nap)3(11)2]·6(BF4) metallo-cryptophane in an ethereal solution of iodine resulted in rapid I2 uptake. Conversely to the syn-[Pd3(Nap)3(13)2]·6(BF4)·3(DCB) assembly, in which the DCB guests were located outside of the cage cavity, single crystal X-ray diffraction analysis showed the inclusion of iodine endo to the cage, where it was non-covalently bound by the tribenzo[a,d,g]cyclononatriene core of the metallo-cryptophane, Fig. 13c. Additional iodine molecules were also observed in the smaller, interstitial sites between the cages at an overall stoichiometry of syn-[Pd3(Nap)3(11)2]·6(BF4)·3(I2). The uptake of iodine could also be effected by subjecting the crystals to an atmosphere of iodine vapours, where they rapidly turned from pale yellow to dark red as the guest was taken up into the crystal lattice. Independent of the route, the uptake of iodine by the syn-[Pd3(Nap)3(11)2]·6(BF4) metallo-cryptophane was irreversible and could not be reversed by solvent-exchange or leaching. This indicates their potential application as sequestering agents for radioactive iodine.74 The uptake of small molecules into a crystal lattice in a SCTSC manner most commonly occurs with porous materials such as metal–organic frameworks, but has been reported for a small number of other systems that are classically nonporous, including organic cage systems,75t-butyl-calix[4]arene,76 and other metallacyclic species.77
2.6 The Pd6L8 stella octangula family
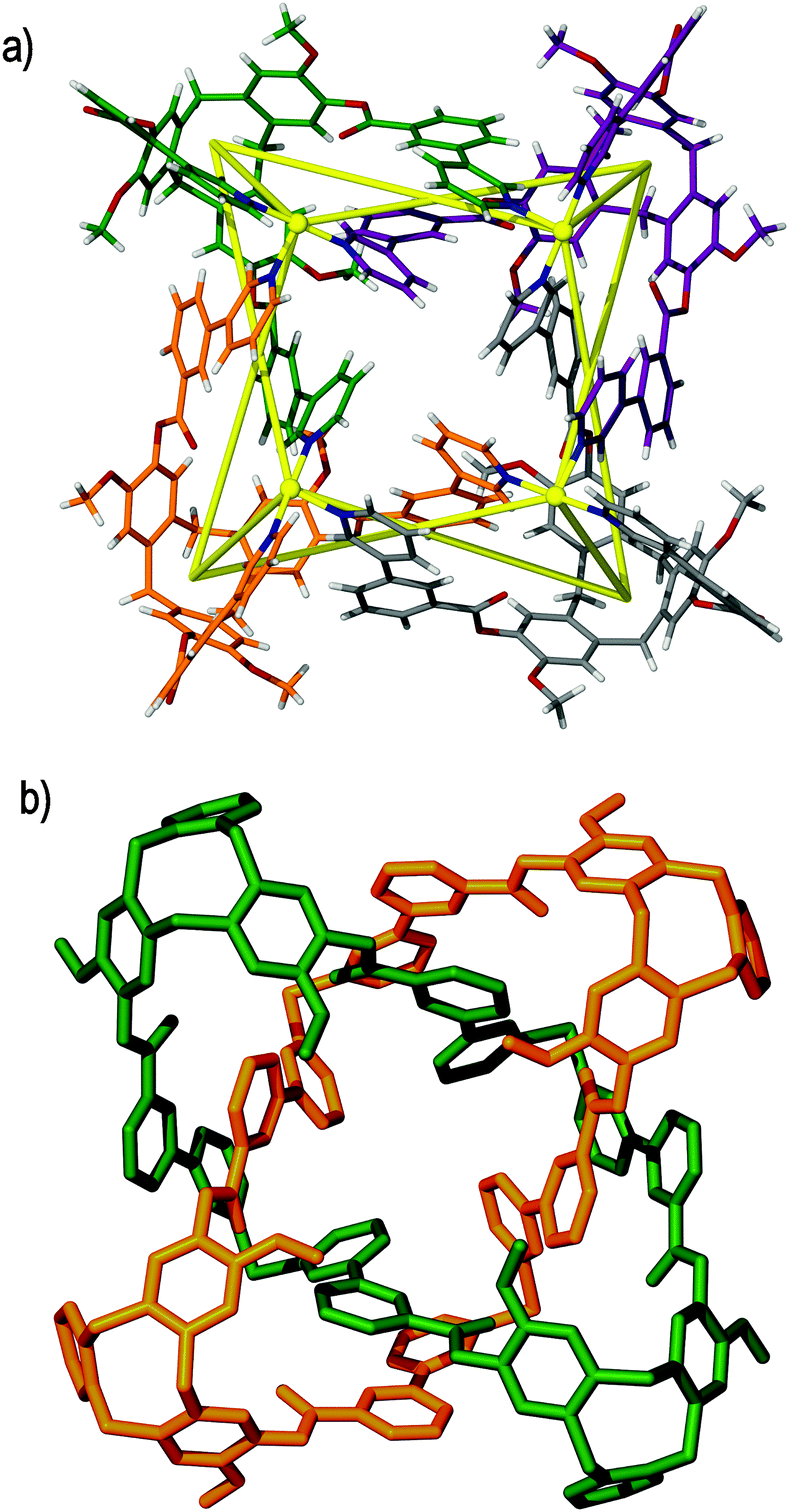
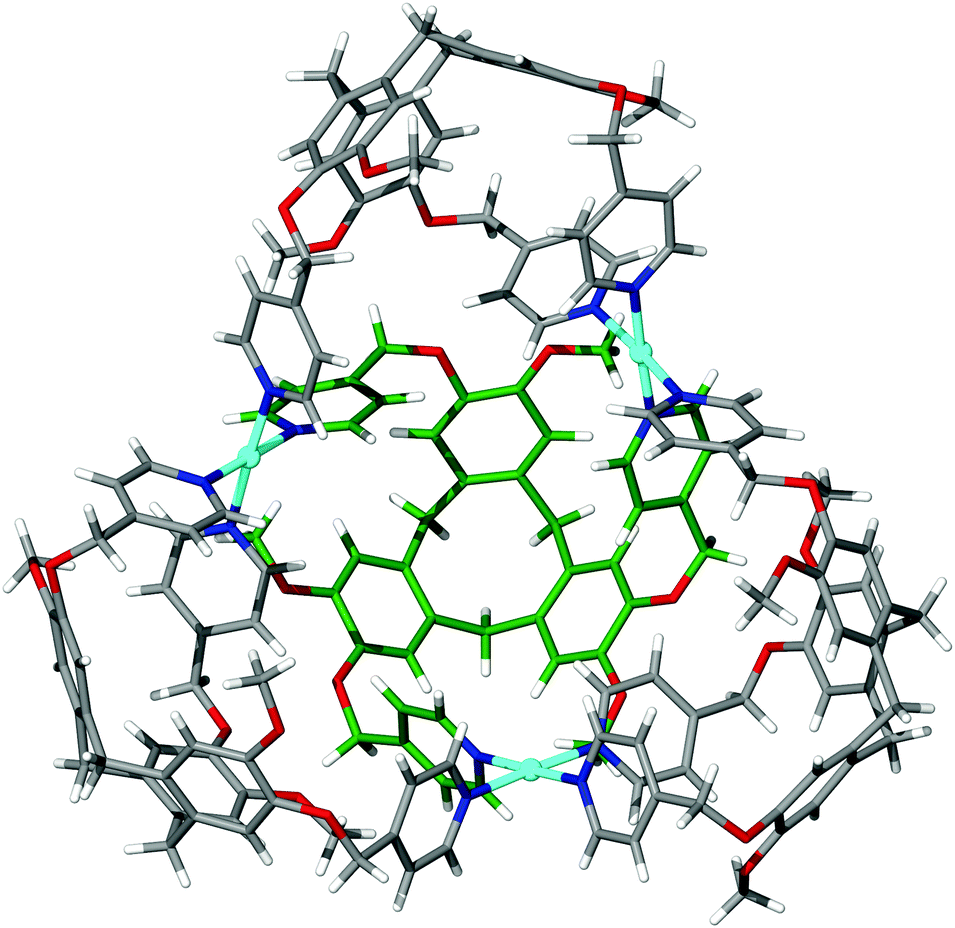
The largest metallo-cages that utilise CTV-type ligands are the [M6L8]12+ stella octangula assemblies, which can be assembled using CTV scaffold ligands that have a tripodal arrangement of 4-pyridyl donor groups. The first reported example was the [Pd6(11)8]12+ ‘stella octangula’, which was accessed from the self-assembly of ligand 11 and Pd(NO3)2 in DMSO solution, and can also be assembled using other Pd(II) salts. The [Pd6(11)8]12+ cage features an octahedral framework of palladium(II) cations that are each coordinated by eight face-capping 11 ligands, Fig. 14a. The resultant coordination cage possesses Oh symmetry and spans 3 nm, in which the inwardly-orientated ligands afford a large, hydrophobic void within the cage scaffold.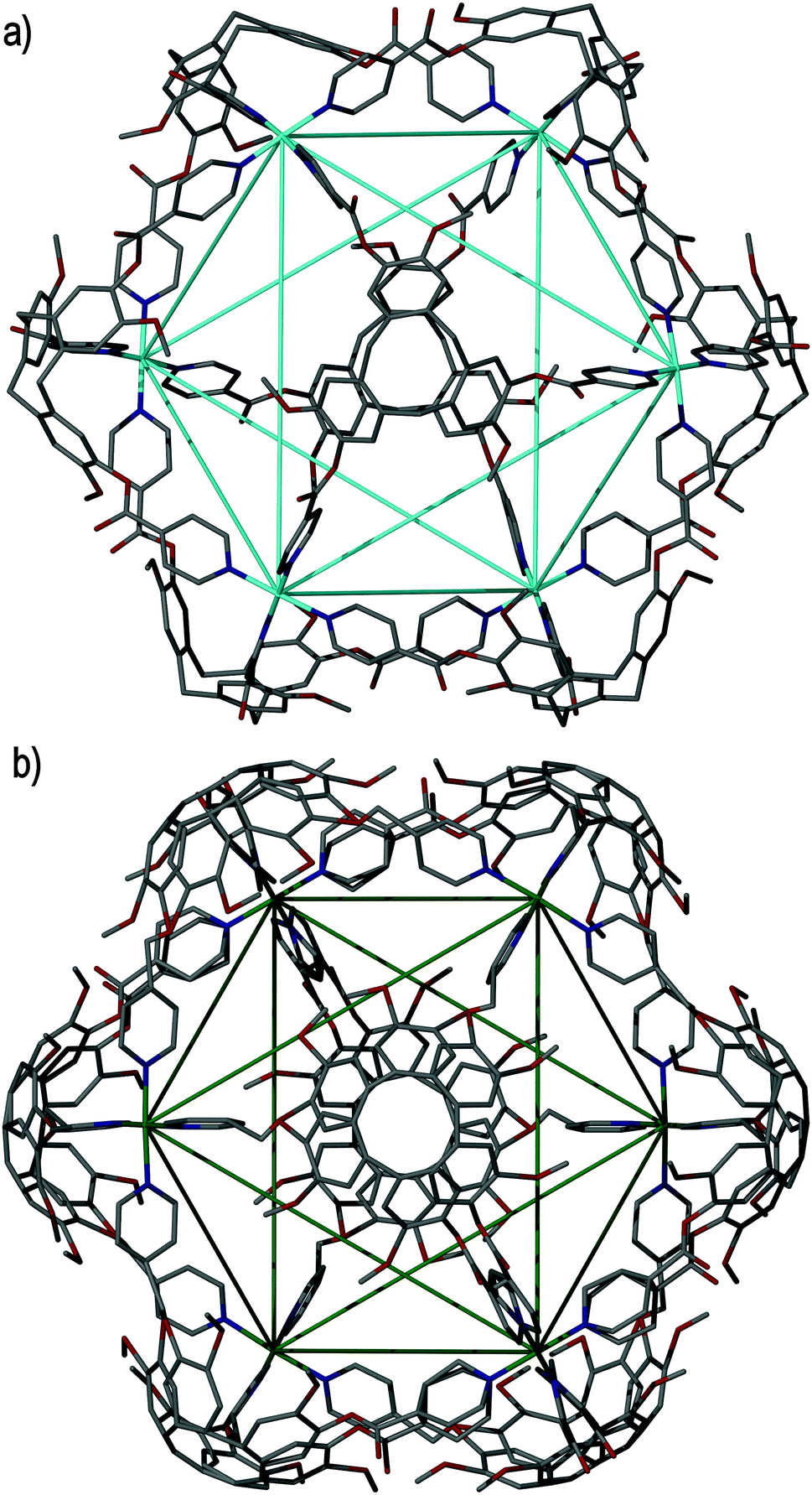 | ||
| Fig. 14 The solid state structures of (a) enantiopure [Pd6(11)8]12+ and (b) mixed enantiomer [Pd6(13)8]12+ ‘stella octangula’ cage assemblies, in which the connectivity of the central octahedral Pd(II) core is depicted using blue and green solid lines, respectively.64,69 | ||
The ‘stella octangula’ moniker is assigned as each face of the octahedral framework is extended out to a single point through ligand coordination, resulting in a spiked appearance that closely resembles the first stellation of an octahedron. Although coordination cages resembling Platonic and Archimedean solids are an increasingly common product of self-assembly, their stellated analogues remain comparatively rare.78 A family of [Pd6L8]12+ stella octangula cages can be assembled with the 4-pyridyl donor ligands 11, 13, 15 and 16. Likewise, the Pt(II) analogue can be accessed using the most soluble of these ligands, the propylated ligand 13; however, unlike for the [Pd6L8]12+ species, the platinum(II) congener [Pt6(13)8]12+ does not form quantitatively. M6L8 coordination cages that utilise achiral tripodal ligands, possessing the same Oh symmetry but without the stellation aspect to their structures, are also known.79
Whilst ligands 11 and 13 are sterically and interactionally equivalent, the self-assembly of the [Pd6(11)8]12+ and [Pd6(13)8]12+ ‘stella octangula’ cages is subtly different. ESI-MS shows that the [Pd6(11)8]12+ and [Pd6(13)8]12+ cages each assemble rapidly and quantitatively in DMSO solution. However, 1H NMR studies reveal that the methylated [Pd6(11)8]12+ cage undergoes further equilibration with time, although with no changes to its stoichiometry or relative size. Interestingly, the [Pd6(13)8]12+ assembly shows no further equilibration with time in DMSO solution. Determination of the crystal structures of each ‘stella octangula’ cage indicates that this further equilibration is due to chiral self-sorting that occurs for the [Pd6(11)8]12+ assembly but not the [Pd6(13)8]12+ assembly, Fig. 14. Each [Pd6(11)8]12+ cage features only a single ligand enantiomer (M or P) about the Pd6 framework, and their overall composition is a racemic mixture of (+)-[Pd6L8]12+ and (−)-[Pd6L8]12+ cages. In comparison, the crystal structure of the [Pd6(13)8]12+ cage shows a disordered assembly comprising a mixture of M and P ligand enantiomers. Thus, it was inferred that the initial [Pd6(11)8]12+ and [Pd6(13)8]12+ assemblies pertained to mixed enantiomer, kinetic products and that the equilibrium process observed for the methylated [Pd6(11)8]12+ assembly represented a thermodynamically-driven ligand exchange towards enantiopurity.80 Interestingly, the propylated [Pd6(13)8]12+ cage displayed self-sorting phenomena in other solvents. Whereas its assembly in DMSO solution afforded only cages of mixed ligand enantiomer, its assembly in acetonitrile and nitromethane solvents facilitated ligand exchange towards enantiopure [Pd6(13)8]12+ ‘stella octangula’ cages.
The methylated [Pd6(11)8]12+ stella octangula cage was observed by Fisher and coworkers to form host–guest complexes with a range of sodium alkyl sulfates in DMSO solution, in which the amphiphilic nature of the surfactant molecules facilitated their inclusion.81 Here, electrostatic interactions between the sulfate head group and cationic cage framework assisted in orientating the guests in such a way that their alkyl chains were able to interact with the hydrophobic interior, driven by the formation of alkyl–alkyl interactions.82 The increased solubility of the propylated [Pd6(13)8]12+ assembly was found to facilitate the formation of inclusion complexes with ortho-carborane guest in the gas-phase.64
Intriguingly, the self-assembly of ligand (±)-tris-(4-pyridylmethyloxy)cyclotriguaiacylene 14 and palladium(II) nitrate does not afford the [Pd6(14)8]12+ stella octangula cage, and instead forms an open [Pd3(14)4]6+ ‘super-bowl’ assembly that can be approximated as half the parent cage, Fig. 15.83 Whereas the [Pd6(11)8]12+ ‘stella octangula’ features eight identical ligands that each coordinate to three different palladium(II) centres, the ligands in the [Pd3(14)4]6+ half-cage display dissimilar coordination environments. The ligand at the base of the assembly coordinates to three crystallographically distinct palladium(II) centres, whilst the other three coordinate to only two. This is likely as a result of increased conformational freedom of ligand 14 over the ester-linked derivative 11 and again supports the notion that ligand rigidity is a principal factor in their predictable assembly. Whilst the resulting assembly may be considered a ‘half-cage’, a pseudo ‘stella octangula’ cage is afforded in the solid state through the hydrogen-bonded dimerisation of two [Pd3(14)4]6+ assemblies.
 | ||
| Fig. 15 The skeletal structure of the [Pd3(14)4]6+ half-cage, closely resembling half of a ‘stella octangula’ cage. The ligand at the base of the tetrameric assembly is coloured green for clarity.83 | ||
2.7 Ligand exchange and ‘stella octangula’ cage dynamics
The ability of ligands 11 and 13 to afford structurally analogous [Pd6(11)8]12+ and [Pd6(13)8]12+ cages allowed for any ligand exchange phenomena84 between homoleptic and heteroleptic ‘stella octangula’ cage mixtures to be probed.64 Interestingly, the preformed and equilibrated [Pd6(11)8]12+ and [Pd6(13)8]12+ assemblies were observed to be indefinitely stable to one another in DMSO solution. In spite of the lability of the Pd–N bond these remained truly homoleptic, with no evidence for ligand exchange occurring over a monitoring period that extended for many months. A statistical mixture of heteroleptic [Pd6(11)n(13)8−n]12+ cages can be formed from the self-assembly of a mixture of ligands 11 and 13 with palladium(II) salts in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:6 ratio. Similarly, once formed these heteroleptic cages did not undergo any further ligand exchange upon prolonged standing.64
:4:6 ratio. Similarly, once formed these heteroleptic cages did not undergo any further ligand exchange upon prolonged standing.64
The homoleptic cages could be perturbed, however, by addition of excess ligand. Whilst the addition of propylated ligand 13 to a preformed and equilibrated [Pd6(11)8]12+ cage had no substantial effect upon the speciation, the addition of methylated ligand 11 to the [Pd6(13)8]12+ cage saw rapid and quantitative ligand exchange in favour of the [Pd6(11)8]12+ assembly, with propylated ligand 13 being displaced into solution. These differences in cage stability allow us establish an assembly–disassembly cycle, in which high fidelity speciation control can be effected, Fig. 16. Treatment of the homoleptic [Pd6(11)8]12+ and [Pd6(13)8]12+ cage mixture with N,N′-dimethylaminopyridine (DMAP) in DMSO solution effected the rapid and quantitative degradation of the homoleptic cages to the mononuclear [Pd(DMAP)4]2+ complex and a mixture of uncoordinated ligands, driven by the strong Lewis basicity of the competing DMAP ligands.85 Treatment of this mixture with p-toluenesulfonic acid (TsOH) selectively protonated the DMAP ligands and thus afforded a mixture of heteroleptic [Pd6(11)n(13)8−n]12+ cages in solution. Following, the addition of ligands 11 and 13 facilitated ligand exchange about the Pd6 cage framework to reform the biased mixture of homoleptic [Pd6(11)8]12+ and [Pd6(13)8]12+ cages, which further equilibrated over time to afford the racemic mixture of enantiopure [Pd6(11)8]12+ and mixed enantiomer [Pd6(13)8]12+ cages.64
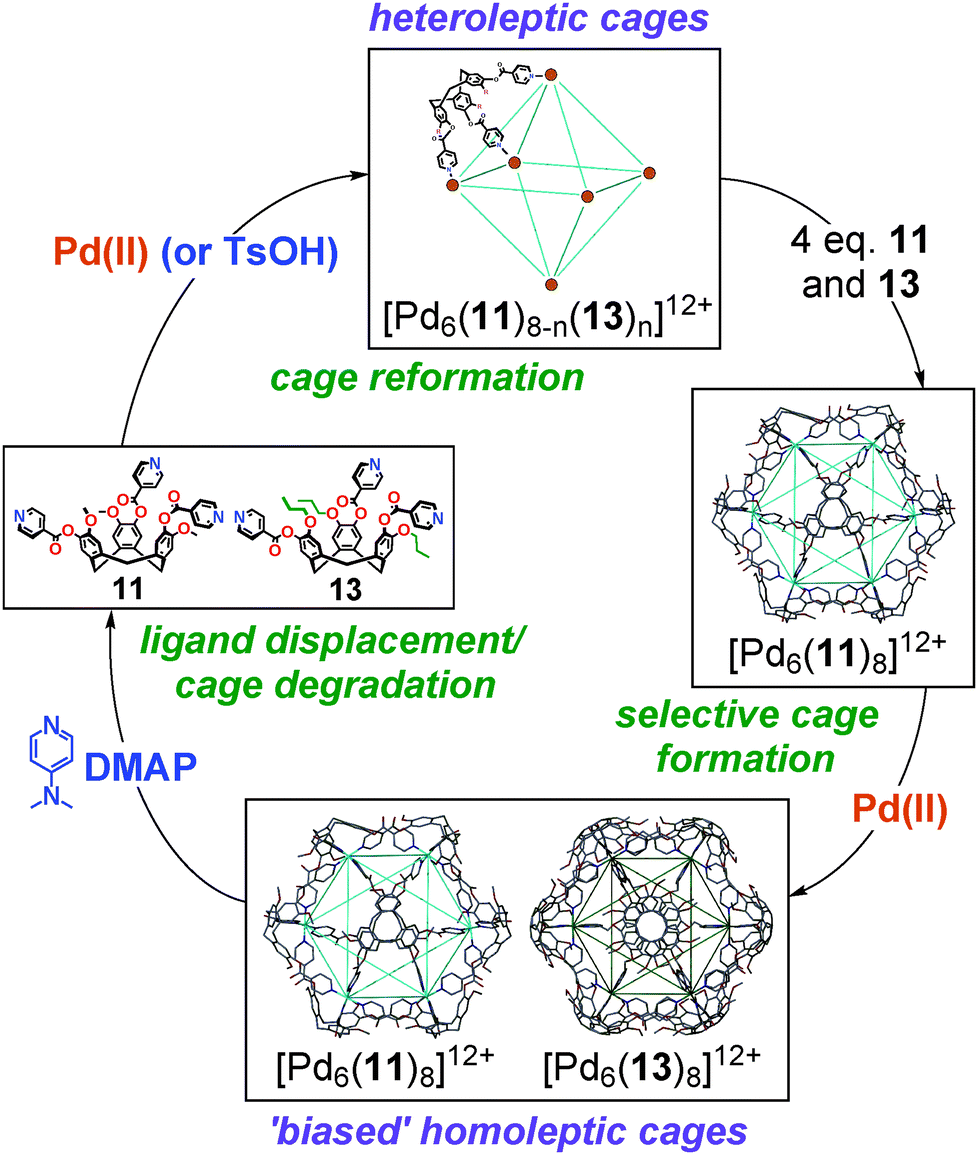 | ||
| Fig. 16 The complex assembly–disassembly cycle determined for the chirality and speciation control experiments between all accessible ‘stella octangula’ coordination cages.64 | ||
The larger [Pd6(15)8]12+ and [Pd6(16)8]12+ assemblies (where 15 and 16 = (±)-tris-(4-(4-pyridyl)benzoyl)cyclotriguaiacylene and (±)-tris-(4-(4-pyridyl)benzoyl)-tris-(propoxy)cyclotricatechylene, respectively) show distinct behavior again. Whilst the heteroleptic [Pd6(15)n(16)8−n]12+ cage mixture was successfully prepared and shown to not further equilibrate with time, a mixture of the homoleptic [Pd6(15)8]12+ and [Pd6(16)8]12+ cages were observed to undergo limited ligand exchange in DMSO solution over a period of months.64 In this instance, the increased conformational flexibility of the larger cages result in considerable structural permutations at the metal coordination geometry that are believed to promote and accelerate ligand exchange.
3 Conclusions and outlook
As was first recognized by Shinkai, Yamaguchi and co-workers, the CTV scaffold represents an attractive framework for the rational design and self-assembly of coordination cages. The pre-organised pyramidal shape means that CTV ligands are predisposed to form cages upon convergent assembly, and metal-binding ligand moieties can be appended to the CTV scaffold in a relatively straightforward manner. Future challenges in this field include establishing “at will” construction of target cages, most particularly an understanding of the formation of unanticipated topologically complicated assemblies, and use of solution phase behaviour to fully develop the known cages as nano-scale vessels.The self-assembly products afforded are in some cases predictable, such as for the formation of Abraham and Robson's [M6L4] tetrahedral cages with deprotonated CTC and in the family of [Pd6L8]12+ ‘stella octangula’ cages. This is certainly not always the case, however, but the serendipitous discoveries along the way are intriguing in their own right, most particularly the formation of the topologically complicated assemblies of the [2]catenane metallo-cryptophanes and the “Solomon's cube”, a self-entangled cube of highly unusual topology. From their isolation it is possible to identify factors that make predicting their assembly challenging. Perhaps unsurprisingly, ligands of increased rigidity behave more predictably than do ligands with a higher degree of flexibility, and increased ligand solubility leads to more predictable behavior. The host functionality of the cavitand can also be a factor, noting that the ‘hand-shake’ self-inclusion motif commonly observed in clathrate complexes of C3-symmetric CTV derivatives also occurs within [Ag4(4)4]4+ tetrahedra. The binding of a bulky guest in the molecular cavity may direct the self-assembly processes towards the formation of a coordination polymer rather than a cage. Curiously, the simplest coordination cage on paper—the M3L2 metallo-cryptophane—has proved to be one of the more challenging to access with our library of ligands, with unanticipated [2]catenane formation, assemblies apparent in the solid state not forming in solution, and M3L2 to M6L8 ligand-exchange rearrangements all occurring. The preparation of stable and well-defined single cage M3L2 metallo-cryptophanes was finally achieved by use of an organometallic cis-protecting ligand, and the crystalline materials thus accessed exhibited the ability to uptake guests in a single-crystal-to-single-crystal manner.
The inherent chirality of tripodal CTV-type ligands means that chiral assemblies can be accessed and a number of systems exhibit chiral self-sorting from an enantiomeric mixture of ligands. The [Pd6L8]12+ stella octangula cages illustrate that cage stability and solution phase behaviour is dependent on the exact ligand and solvent used. This affords the ability to selectively control the speciation and overall chirality of cage mixtures in solution with high fidelity, which is an unusual property that may have future application in the chiral discrimination of guest molecules. Likewise, the ability to cyclically assemble and disassemble a coordination cage at will has potential application in cargo delivery.
Acknowledgements
The authors thank the EPSRC, Charles Brotherton Trust (JJH) and the University of Leeds for funding much of the research described herein, and the STFC for access to beamline facilities. We thank collaborators and co-workers whose work is cited herein, including Julie Fisher, Lindsay P. Harding, Andrew I. Cooper, Rob Clowes, Malcolm Halcrow, Christopher J. Sumby, Tanya K. Ronson, Aleema Westcott, Christopher Carruthers, Marc A. Little, Scott E. Chambers, Nikki J. Cookson, Jessica Donkin and Jordan Loder.Notes and references
- For examples and reviews, see: (a) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (b) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (c) K. Harris and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (e) M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC; (f) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed.
- (a) M. J. Hardie, Chem. Soc. Rev., 2010, 39, 516–527 RSC; (b) J. W. Steed, H. Zhang and J. L. Atwood, Supramol. Chem., 1996, 7, 37–45 CrossRef CAS PubMed; (c) A. Collet, Tetrahedron, 1987, 43, 5725–5759 CrossRef CAS.
- (a) J. L. Atwood, M. J. Barnes, M. G. Gardiner and C. L. Raston, Chem. Commun., 1996, 1449–1450 RSC; (b) J. W. Steed, P. C. Junk, J. L. Atwood, M. J. Barnes, C. L. Raston and R. S. Burkhalter, J. Am. Chem. Soc., 1994, 116, 10346–10347 CrossRef CAS; (c) E. Huerta, G. A. Metselaar, A. Fragoso, E. Santos, C. Bo and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 202–205 CrossRef CAS PubMed; (d) E. Huerta, E. Cequier and J. de Mendoza, Chem. Commun., 2007, 5016–5018 RSC; (e) E. Huerta, H. Isla, E. M. Perez, C. Bo, N. Martin and J. de Mendoza, J. Am. Chem. Soc., 2010, 132, 5351–5353 CrossRef CAS PubMed.
- (a) C. Carruthers, J. Fisher, L. P. Harding and M. J. Hardie, Dalton Trans., 2010, 39, 355–357 RSC; (b) R. J. Blanch, M. Williams, G. D. Fallon, M. G. Gardiner, R. Kaddour and C. L. Raston, Angew. Chem., Int. Ed. Engl., 1997, 36, 504–506 CrossRef CAS PubMed.
- (a) D. J. Cram, Angew. Chem., Int. Ed., 1988, 27, 1009–1020 CrossRef PubMed; (b) D. J. Cram, Nature, 1992, 356, 29–36 CrossRef CAS PubMed; (c) R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed; (d) S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC; (e) S. Kennedy, G. Karotsis, C. M. Beavers, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Angew. Chem., Int. Ed., 2010, 49, 4205–4208 CrossRef CAS PubMed; (f) S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef PubMed; (g) G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049–1052 CrossRef CAS; (h) H. Zhang and Y. Zhao, Chem. – Eur. J., 2013, 19, 16862–16879 CrossRef CAS PubMed.
- (a) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS PubMed; (b) J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed; (c) A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC; (d) C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1992, 31, 528–549 CrossRef PubMed.
- For examples and reviews, see: (a) Y. Bi, S. Du and W. Liao, Coord. Chem. Rev., 2015, 276, 61–72 CrossRef PubMed; (b) R. Pinalli, F. Boccini and E. Dalcanale, Isr. J. Chem., 2011, 51, 781–797 CrossRef CAS PubMed; (c) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760–1768 CrossRef CAS PubMed; (d) T. Haino, M. Kobayashi and Y. Fukuzawa, Chem. – Eur. J., 2006, 12, 3310–3319 CrossRef CAS PubMed; (e) H. Jude, D. J. Sinclair, N. Das, M. S. Sherburn and P. J. Stang, J. Org. Chem., 2006, 71, 4155–4163 CrossRef CAS PubMed; (f) O. D. Fox, J. Cookson, E. J. S. Wilkinson, M. G. B. Drew, E. J. MacLean, S. J. Teat and P. D. Beer, J. Am. Chem. Soc., 2006, 128, 6990–7002 CrossRef CAS PubMed; (g) R. M. McKinlay, G. W. V. Cave and J. L. Atwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5944–5948 CrossRef CAS PubMed; (h) M. Yamanaka, Y. Yamada, Y. Sei, K. Yamaguchi and K. Koyayashi, J. Am. Chem. Soc., 2006, 128, 1531 CrossRef CAS PubMed; (i) E. Menozzi, M. Busi, R. Ramingo, M. Campagnolo, S. Geremia and E. Dalcanale, Chem. – Eur. J., 2005, 11, 3136–3148 CrossRef CAS PubMed; (j) K. Kobayashi, Y. Yamada, M. Yamanaka, Y. Sei and K. Yamaguchi, J. Am. Chem. Soc., 2004, 126, 13896–13897 CrossRef CAS PubMed; (k) L. Baldini, P. Ballester, A. Casnati, R. M. Gomila, C. A. Hunter, F. Sansone and R. Ungaro, J. Am. Chem. Soc., 2003, 125, 14181–14189 CrossRef CAS PubMed; (l) O. D. Fox, N. K. Dalley and R. G. Harrison, J. Am. Chem. Soc., 1998, 120, 7111–7112 CrossRef CAS.
- (a) M. J. Hardie and C. L. Raston, Cryst. Growth Des., 2001, 1, 53–58 CrossRef CAS; (b) D. V. Konarev, S. S. Khasanov, I. I. Vorontsov, G. Saito, M. Yu. Antpin, A. Otsuka and R. N. Lyubovskaya, Chem. Commun., 2002, 2548–2549 RSC; (c) R. Ahmad, I. Dix and M. J. Hardie, Inorg. Chem., 2003, 42, 2182–2184 CrossRef CAS PubMed; (d) R. Ahmad, A. Franken, J. D. Kennedy and M. L. Hardie, Chem. – Eur. J., 2004, 10, 2190–2198 CrossRef CAS PubMed; (e) S. Tashiro, S. Hashida and M. Shionoya, Chem. – Asian J., 2012, 7, 1180–1184 CrossRef CAS PubMed.
- (a) J. Canceill, J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1983, 122–123 RSC; (b) J. J. Loughrey, C. A. Kilner, M. J. Hardie and M. A. Halcrow, Supramol. Chem., 2011, 24, 2–13 CrossRef PubMed; (c) M. A. Little, J. J. Loughrey, A. Santoro, M. A. Halcrow and M. J. Hardie, Tetrahedron Lett., 2014, 55, 2530–2533 CrossRef CAS PubMed; (d) C. Garcia, J. Malthete and A. Collet, Bull. Soc. Chim. Fr., 1993, 93–95 CAS; (e) M. J. Hardie, R. Ahmad and C. J. Sumby, New J. Chem., 2005, 29, 1231–1240 RSC.
- (a) M. J. Hardie, R. M. Mills and C. J. Sumby, Org. Biomol. Chem., 2004, 2, 2958–2964 RSC; (b) M. J. Hardie and C. J. Sumby, Inorg. Chem., 2004, 43, 6872–6874 CrossRef CAS PubMed.
- A. Collet, J. Gabard, J. Jacques, M. Cesario, J. Guilhem and C. Pascard, J. Chem. Soc., Perkin Trans. 1, 1981, 1630–1638 RSC.
- H. Zimmermann, P. Tolstoy, H.-H. Limbach, R. Poupko and Z. Luz, J. Phys. Chem. B, 2004, 108, 18772–18778 CrossRef CAS.
- O. Lafon, P. Lesot, H. Zimmermann, R. Poupko and Z. Luz, J. Phys. Chem. B, 2007, 111, 9453–9467 CrossRef CAS PubMed.
- J. Costante, C. Garcia and A. Collet, Chirality, 1997, 9, 446–453 CrossRef CAS.
- J. Canceill, A. Collet, J. Gabard, G. Gottarelli and G. P. Spada, J. Am. Chem. Soc., 1985, 107, 1299–1308 CrossRef CAS.
- (a) T. Brotin and J.-P. Dutasta, Chem. Rev., 2008, 109, 88–130 CrossRef PubMed; (b) K. T. Holman, Encyclopedia of Supramolecular Chemistry, CRC Press, 2004, pp. 340–348 Search PubMed.
- O. Taratula, P. A. Hill, N. S. Khan, P. J. Carroll and I. J. Dmochowski, Nat. Commun., 2010, 1, 148 CrossRef PubMed.
- H.-R. Tseng, S. A. Vignon, P. C. Celestre, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem. – Eur. J., 2003, 9, 543–556 CrossRef CAS PubMed.
- M. A. Little, J. Donkin, J. Fisher, M. A. Halcrow, J. Loder and M. J. Hardie, Angew. Chem., Int. Ed., 2011, 51, 764–766 CrossRef PubMed.
- (a) L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef PubMed; (b) P. Berthault, H. Desvaux, T. Wendlinger, M. Gyejacquot, A. Stopin, T. Brotin, J.-P. Dutasta and Y. Boulard, Chem. – Eur. J., 2010, 16, 12941–12946 CrossRef CAS PubMed.
- C. Givelet, J. Sun, D. Xu, T. J. Emge, A. Dhokte and R. Warmuth, Chem. Commun., 2011, 47, 4511–4513 RSC.
- R. M. Fairchild and K. T. Holman, J. Am. Chem. Soc., 2005, 127, 16364–16365 CrossRef CAS PubMed.
- (a) P. D. Raytchev, O. Perraud, C. Aronica, A. Martinez and J.-P. Dutasta, J. Org. Chem., 2010, 75, 2099–2102 CrossRef CAS PubMed; (b) B. Chatelet, E. Payet, O. Perraud, P. Dimitrov-Raytchev, L.-L. Chapellet, V. Dufaud, A. Martinez and J.-P. Dutasta, Org. Lett., 2011, 13, 3706–3709 CrossRef CAS PubMed.
- D. Xu and R. Warmuth, J. Am. Chem. Soc., 2008, 130, 7520–7521 CrossRef CAS PubMed.
- M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed.
- R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS PubMed.
- (a) M. A. Little, T. K. Ronson and M. J. Hardie, Dalton Trans., 2011, 40, 12217–12227 RSC; (b) T. K. Ronson and M. J. Hardie, CrystEngComm, 2008, 10, 1731–1734 RSC; (c) S. T. Mough and K. T. Holman, Chem. Commun., 2008, 1407–1409 RSC.
- K. T. Holman, M. M. Halihan, S. S. Jurisson, J. L. Atwood, R. S. Burkhalter, A. R. Mitchell and J. W. Steed, J. Am. Chem. Soc., 1996, 118, 9567–9576 CrossRef CAS.
- R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS PubMed.
- D. S. Bohle and D. Stasko, Chem. Commun., 1998, 567–569 RSC.
- (a) B. F. Abrahams, N. J. FitzGerald and R. Robson, Angew. Chem., Int. Ed., 2010, 49, 2896–2899 CrossRef CAS PubMed; (b) B. F. Abrahams, B. A. Boughton, N. J. FitzGerald, J. L. Holmes and R. Robson, Chem. Commun., 2011, 47, 7404–7406 RSC.
- M. J. Hardie, Isr. J. Chem., 2011, 51, 807–816 CrossRef CAS PubMed.
- J. J. Henkelis, S. A. Barnett, L. P. Harding and M. J. Hardie, Inorg. Chem., 2012, 51, 10657–10674 CrossRef CAS PubMed.
- J. J. Henkelis and M. J. Hardie, CrystEngComm, 2014, 16, 8138–8146 RSC.
- M. A. Little, M. A. Halcrow, L. P. Harding and M. J. Hardie, Inorg. Chem., 2010, 49, 9486–9496 CrossRef CAS PubMed.
- M. A. Little, M. A. Halcrow and M. J. Hardie, Chem. Commun., 2013, 49, 1512–1514 RSC.
- C. J. Sumby and M. J. Hardie, Angew. Chem., Int. Ed., 2005, 44, 6395–6399 CrossRef CAS PubMed.
- (a) A. Martinez, L. Guy and J.-P. Dutasta, J. Am. Chem. Soc., 2010, 132, 16733–16734 CrossRef CAS PubMed; (b) C. Browne, S. Brenet, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 125, 1998–2002 CrossRef PubMed; (c) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744–2751 RSC; (d) K. Suzuki, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 2819–2822 CrossRef CAS PubMed.
- For example (a) P. Kruger, A. Ferguson, M. A. Squire, R. Clerac, C. Mathoniere, D. Mitcov and D. Siretanu, Chem. Commun., 2013, 49, 1597–1599 RSC; (b) S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed; (c) Y. Jiao, J. Wang, P. Wu, L. Zhao, C. He, J. Zhang and C. Duan, Chem. – Eur. J., 2014, 20, 2224–2231 CrossRef CAS PubMed; (d) A. Jiménez, R. A. Bilbeisi, T. K. Ronson, S. Zarra, C. Woodhead and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 4556–4560 CrossRef PubMed.
- C. J. Sumby, M. J. Carr, A. Franken, J. D. Kennedy, C. A. Kilner and M. J. Hardie, New J. Chem., 2006, 30, 1390–1396 RSC.
- C. J. Sumby, J. Fisher, T. J. Prior and M. J. Hardie, Chem. – Eur. J., 2006, 12, 2945–2959 CrossRef CAS PubMed.
- C. Carruthers, T. K. Ronson, C. J. Sumby, A. Westcott, L. P. Harding, T. J. Prior, P. Rizkallah and M. J. Hardie, Chem. – Eur. J., 2008, 14, 10286–10296 CrossRef CAS PubMed.
- T. K. Ronson, J. Fisher, L. P. Harding, P. J. Rizkallah, J. E. Warren and M. J. Hardie, Nat. Chem., 2009, 1, 212–216 CrossRef CAS PubMed.
- (a) J. E. Beves, C. J. Campbell, D. A. Leigh and R. G. Pritchard, Angew. Chem., Int. Ed., 2013, 52, 6464–6467 CrossRef CAS PubMed; (b) C. D. Pentecost, K. S. Chichak, A. J. Peters, G. W. V. Cave, S. J. Cantrill and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 218–222 CrossRef CAS PubMed; (c) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2012, 42, 1700–1712 RSC.
- Z. Zhong, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Org. Lett., 2001, 3, 1085–1087 CrossRef CAS PubMed.
- For example (a) U. Radhakrishnan, M. Schweiger and P. J. Stang, Org. Lett., 2001, 3, 3141–3143 CrossRef CAS PubMed; (b) C. G. Claessens and T. Torres, Chem. Commun., 2004, 1298–1299 RSC; (c) H.-B. Yang, K. Ghosh, A. M. Arif and P. J. Stang, J. Org. Chem., 2006, 71, 9464–9469 CrossRef CAS PubMed; (d) S. Tartaggia, O. De Lucchi, A. Gambaro, R. Zangrando, F. Fabris and A. Scarso, Chem. – Eur. J., 2013, 19, 5701–5714 CrossRef CAS PubMed.
- J. J. Henkelis, T. K. Ronson, L. P. Harding and M. J. Hardie, Chem. Commun., 2011, 47, 6560–6562 RSC.
- (a) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (b) C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- C. Piguet, Dalton Trans., 2011, 40, 8059–8071 RSC.
- S.-K. Lin, J. Chem. Inf. Comput. Sci., 1996, 36, 367–376 CrossRef CAS.
- (a) D. R. Link, G. Natale, R. Shao, J. E. Maclennan, N. A. Clark, E. Körblova and D. M. Walba, Science, 1997, 278, 1924–1927 CrossRef CAS; (b) J.-T. Yu, Y.-Y. Shi, J. Sun, J. Lin, Z.-T. Huang and Q.-Y. Zheng, Sci. Rep., 2013, 3, 2947 Search PubMed; (c) K. K. Bisht and E. Suresh, J. Am. Chem. Soc., 2013, 135, 15690–15693 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Jr., Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950–2951 CrossRef CAS PubMed.
- K. Mislow and R. Bolstad, J. Am. Chem. Soc., 1955, 77, 6712–6713 CrossRef CAS.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS.
- (a) R. Zhu, J. Luebben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 59, 2796–2800 CrossRef PubMed; (b) A. Mishra, A. Dubey, J. W. Min, H. Kim, P. J. Stang and K.-W. Chi, Chem. Commun., 2014, 50, 7542–7544 RSC; (c) S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauedorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed; (d) S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed; (e) A. K. Bar, S. Raghothama, D. Moon and P. S. Mukherjee, Chem. – Eur. J., 2012, 18, 3199–3309 CrossRef CAS PubMed; (f) Y. Li, K. M. Mullen, T. D. W. Claridge, P. J. Costa, V. Felix and P. D. Beer, Chem. Commun., 2009, 7134–7136 RSC; (g) M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 706–710 CrossRef CAS PubMed.
- (a) G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1516–1520 CrossRef CAS PubMed; (b) T. Hasell, X. Wu, T. A. Jones, J. Basca, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed.
- T. K. Ronson, H. Nowell, A. Westcott and M. J. Hardie, Chem. Commun., 2010, 47, 176–178 RSC.
- (a) G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285–8288 CrossRef CAS PubMed; (b) J. J. Henkelis, L. F. Jones, M. P. de Miranda, C. A. Kilner and M. A. Halcrow, Inorg. Chem., 2010, 49, 11127–11132 CrossRef CAS PubMed.
- J.-T. Yu, J. Sun, Z.-T. Huang and Q.-Y. Zheng, CrystEngComm, 2012, 14, 112–115 RSC.
- J. J. Henkelis, T. K. Ronson and M. J. Hardie, CrystEngComm, 2014, 16, 3688–3693 RSC.
- J. Wei, Z.-M. Li, X.-J. Jin, X.-J. Yao, X.-P. Cao, H.-F. Chow and D. Kuck, Chem. – Asian J., 2015, 10, 1150–1158 CrossRef CAS PubMed.
- J. J. Henkelis, J. Fisher, S. L. Warriner and M. J. Hardie, Chem. – Eur. J., 2014, 20, 4117–4125 CrossRef CAS PubMed.
- (a) B. Brusilowskij, S. Neubacher and C. A. Schalley, Chem. Commun., 2009, 785–787 RSC; (b) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2010, 133, 858–870 CrossRef PubMed.
- D. K. Chand, R. Manivannan, H. S. Sahoo and K. Jeyakumar, Eur. J. Inorg. Chem., 2005, 3346–3352 CrossRef CAS PubMed.
- (a) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS PubMed; (b) B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS PubMed; (c) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- C. Piguet, Chem. Commun., 2010, 46, 6209–6231 RSC.
- T. K. Ronson, J. Fisher, L. P. Harding and M. J. Hardie, Angew. Chem., Int. Ed., 2007, 46, 9086–9088 CrossRef CAS PubMed.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 1999, 100, 39–92 CrossRef PubMed; (b) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed; (c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (d) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2009, 110, 1746–1787 CrossRef PubMed.
- (a) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (b) M. Rubio, E. Jellema, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Dalton Trans., 2009, 8970–8976 RSC; (c) M. Rubio, M. A. Siegler, A. L. Spek and J. N. H. Reek, Dalton Trans., 2010, 39, 5432–5435 RSC; (d) C. Radloff, F. E. Hahn, T. Pape and R. Frohlich, Dalton Trans., 2009, 7215–7222 RSC.
- (a) N. Sinha, F. Roelfes, A. Hepp, C. Mejuto, E. Peris and F. E. Hahn, Organometallics, 2014, 33, 6898–6904 CrossRef CAS; (b) X.-Q. Xiao, A.-Q. Jia, Y.-J. Lin and G.-X. Jin, Organometallics, 2010, 29, 4842–4848 CrossRef CAS.
- J. J. Henkelis, C. J. Carruthers, S. E. Chambers, R. Clowes, A. I. Cooper, J. Fisher and M. J. Hardie, J. Am. Chem. Soc., 2014, 136, 14393–14396 CrossRef CAS PubMed.
- (a) D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS PubMed; (b) G. Massasso, J. Long, J. Haines, S. Devautour-Vinot, G. Maurin, A. Grandjean, B. Onida, B. Donnadieu, J. Larionova, C. Guérin and Y. Guari, Inorg. Chem., 2014, 53, 4269–4271 CrossRef CAS PubMed; (c) X. Liu, S. A. Y. Zhang, Z. Li, H. Xia, M. Xue and Y. Mu, Chem. Commun., 2014, 50, 8495–8498 RSC.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- (a) J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS PubMed; (b) J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed.
- (a) T. Jacobs and L. J. Barbour, CrystEngComm, 2013, 15, 1512–1514 RSC; (b) T. Jacobs, G. O. Lloyd, J. A. Gertenbach, K. K. Müller-Nedebock, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 4913 CrossRef CAS PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330–333 CrossRef CAS PubMed.
- (a) D. K. Chand, K. Biradha, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2002, 2486–2487 RSC; (b) X.-J. Li, F.-L. Jiang, M.-Y. Wu, S.-Q. Zhang, Y.-F. Zhou and M.-C. Hong, Inorg. Chem., 2012, 51, 4116–4122 CrossRef CAS PubMed; (c) M. B. Duriska, S. M. Neville, J. Lu, S. S. Tremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8919–8922 CrossRef CAS PubMed; (d) S. Hiraoka, K. Harano, M. Shiro, Y. Ozawa, N. Yasuda, K. Toriumi and M. Shionoya, Angew. Chem., Int. Ed., 2006, 45, 6488–6491 CrossRef CAS PubMed; (e) D. Moon, S. Kang, J. Park, K. Lee, R. P. John, H. Won, G. H. Seong, Y. S. Kim, G. H. Kim, H. Rhee and M. S. Lah, J. Am. Chem. Soc., 2006, 128, 3530–3531 CrossRef CAS PubMed.
- (a) Y.-R. Zheng and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 3487–3489 CrossRef CAS PubMed; (b) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- N. J. Cookson, J. J. Henkelis, R. J. Ansell, C. W. G. Fishwick, M. J. Hardie and J. Fisher, Dalton Trans., 2014, 43, 5657–5661 RSC.
- L. Yang, C. Adam, G. S. Nichol and S. L. Cockroft, Nat. Chem., 2013, 5, 1006–1010 CrossRef CAS PubMed.
- T. K. Ronson, C. Carruthers, J. Fisher, T. Brotin, L. P. Harding, P. J. Rizkallah and M. J. Hardie, Inorg. Chem., 2010, 49, 675–685 CrossRef CAS PubMed.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC.
- (a) J. D. Crowley, D. A. Leigh, P. J. Lusby, R. T. McBurney, L.-E. Perret-Aebi, C. Petzold, A. M. Z. Slawin and M. D. Symes, J. Am. Chem. Soc., 2007, 129, 15085–15090 CrossRef CAS PubMed; (b) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
Footnote |
| † Current address: Department of Chemistry, Northwestern University, Evanston, Illinois, 60208, USA. |
| This journal is © The Royal Society of Chemistry 2015 |