
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced electron transfer in a supramolecular triad produced by porphyrin anion-induced electron transfer from tetrathiafulvalene calix[4]pyrrole to Li+@C60†
Christina M.
Davis
a,
Kei
Ohkubo
bc,
Aaron D.
Lammer
a,
Dong Sub
Kim
a,
Yuki
Kawashima
b,
Jonathan L.
Sessler
*a and
Shunichi
Fukuzumi
*bcd
aDepartment of Chemistry, The University of Texas, 105 E. 24th Street-Stop A5300, Austin, Texas 78712-1224, USA. E-mail: sessler@cm.utexas.edu
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University and ALCA and SENTAN, Japan Science and Technology Agency (JST), 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
cDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
dFaculty of Science and Technology, Meijo University and ALCA and SENTAN, Japan Science and Technology Agency (JST), Tempaku, Nagoya, Aichi 468-8502, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp
First published on 7th May 2015
Abstract
Binding of a porphyrin carboxylate anion (1) to tetrathiafulvalene calix[4]pyrrole (TTF-C4P) results in electron transfer from TTF-C4P to Li+@C60 to produce the charge-separated state (1/TTF-C4P˙+/Li+@C60˙−) in benzonitrile. Upon photoexcitation of 1, photoinduced electron transfer from the triplet excited state of 1 to TTF-C4P˙+ occurs to produce the higher energy charge-separated state (1˙+/TTF-C4P/Li+@C60˙−), which decays to the ground state with a lifetime of 4.8 μs.
Three component supramolecular systems that can act as photoactive electron transfer (ET) systems are rare. In fact, we are unaware of any systems wherein three discrete redox active components are brought together through purely non-covalent interactions. Such constructs, to the extent they could be assembled, would provide an interesting complement to covalently linked ensembles or those involving a mixture of covalent and noncovalent linkages. Such putative self-assembled, three-component systems are also inherently rich in “molecular information” in that they might respond to a variety of different chemical stimuli. In principle, this would allow control of the supramolecular ET ensemble. Such assemblies are also of interest because they may allow a greater understanding of the determinants needed to create advanced molecular logic devices where an important readout parameter might be a photoinduced electron transfer (PET) event.1 Here we report a three-component supramolecular “triad” based on hydrogen bond anion recognition and π–π donor–acceptor interactions. This system builds on two previous findings, namely that (1) thermal ET occurs from TTF-calix[4]pyrrole (TTF = tetrathiafulvalene) to lithium-encapsulated fullerene and (2) that photoinduced ET may be triggered by irradiating a TTF-calix[4]pyrrole-porphyrin carboxylate ensemble.2,3
Extensive efforts have so far been devoted to the design and synthesis of covalently linked electron donor–acceptor ensembles, including dyads, triads, tetrads and pentads, many of which have been shown to mimic the energy-transfer and electron-transfer processes seen in biological photosynthetic reaction centers.4–10 In this context, the use of non-covalent interactions, such as metal–ligand coordination, electrostatic effects and hydrogen bonds, has garnered considerable attention because they allow electron transfer donor–acceptor supramolecular dyads to be produced via self-assembly.3,10–14 A current challenge involves extending the chemistry of supramolecular dyads to create higher order constructs, such as supramolecular triads.15 To our knowledge, no reports have appeared that describe supramolecular electron donor–acceptor ensembles composed of three different components, perhaps because most non-covalent interactions are too weak to assemble three components at the same time.16
To address this challenge, we have created a supramolecular triad composed of a porphyrin anion (1), the radical cation of tetrathiafulvalene calix[4]pyrrole (TTF-C4P), and the radical anion of Li+-encapsulated C60 (Li+@C60) (Fig. 1). These components bind to one another in a specific fashion via a combination of electrostatic and donor–acceptor interactions as shown in Fig. 2. We have examined the formation of the triad (1/TTF-C4P˙+/Li+@C60˙−) by monitoring porphyrin anion-induced electron transfer from TTF-C4P to Li+@C60 in benzonitrile (PhCN). We also report the photodynamics of the supramolecular triad.
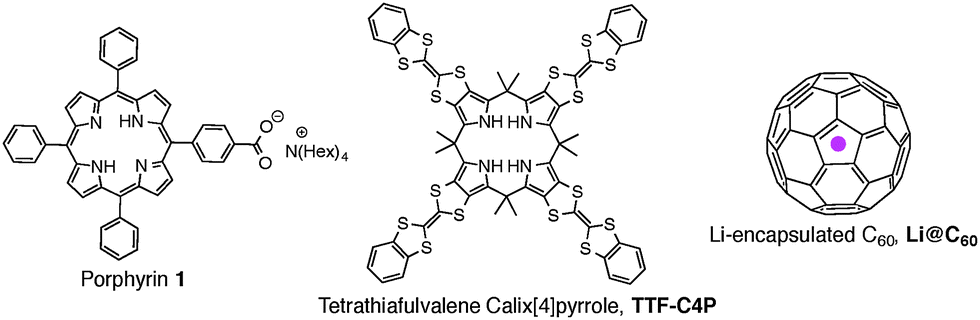 | ||
| Fig. 1 Structures of porphyrin salt 1, tetrathiafulvalene calix[4]pyrrole (TTF-C4P), and Li+@C60. | ||
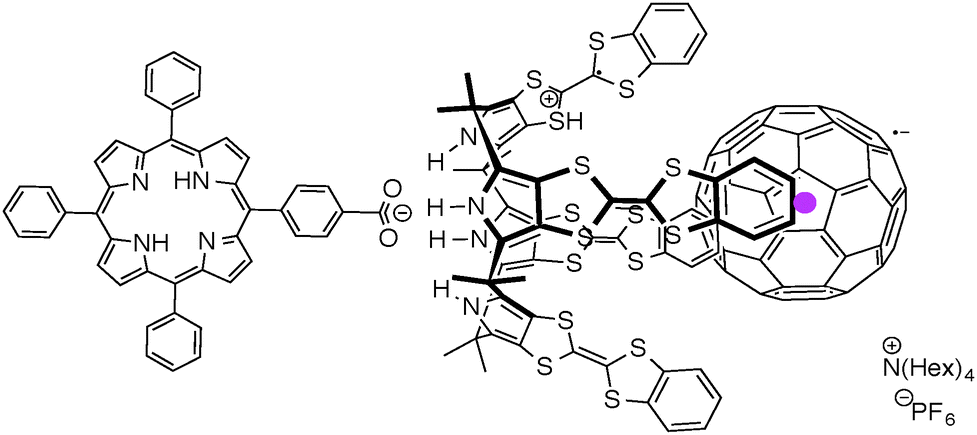 | ||
| Fig. 2 Supramolecular triad composed of porphyrin carboxylate (1), TTF-C4P radical cation, and Li+@C60 radical anion. | ||
Upon mixing TTF-C4P and [Li+@C60]PF6 in PhCN, no evidence of electron transfer was observed. This is consistent with an endergonic electron transfer process as inferred from the higher one-electron oxidation potential of TTF-C4P (Eox = 0.51 V vs. SCE)2,16 compared to the one-electron reduction potential of Li+@C60 (0.14 V vs. SCE).17 Upon addition of tetra-n-hexylammonium (THA+) porphyrin carboxylate (1) to a PhCN solution of TTF-C4P and Li+@C60, however, electron transfer from TTF-C4P and Li+@C60 occurs to produce TTF-C4P˙+ and Li+@C60˙−, as reflected by the appearance of an absorption band at 900 nm due to TTFC4P˙+ and one at 1035 nm due to Li+@C60˙− (see Fig. 3a).1,15,16 The absorbance at both 900 and 1035 nm increased with increasing concentration of 1 to reach a constant value at the point where electron transfer is deemed to be complete (ca. 1 equiv.) as shown in Fig. 3b. The subsequent decrease in the absorption at 1035 nm in Fig. 3b is attributed to competition for the neutral Li+@C60˙− binding site from the THA+ counter cation of the excess porphyrin carboxylate, 1. This competition has a substantial effect on the electron transfer chemistry that gives rise to the long wavelength optical feature (i.e., formation of Li+@C60˙−).
 | ||
| Fig. 3 (a) VIS-NIR spectral changes consistent with electron transfer from TTF-C4P (100 μM) to Li+@C60 (100 μM) as seen in the presence of increasing concentrations of 1 in PhCN. Inset: expanded absorption from 600 to 1600 nm. (b) Plots of absorbance at 1035 nm and 680 nm vs. concentration of 1. | ||
It is known that Cl− binds to TTF-C4P and induces a conformational change from the so-called 1,3-alternate to the cone conformation due to concerted NH-anion hydrogen bonding interactions.2,16 Under conditions of stoichiometric concentrations of 1 or in its absence, such a conformational change makes it possible to bind the fullerene substrate and stabilize the radical ion pair involving the TTF-C4P˙+ host and the bound Li+@C60˙− guest. The observation of effective ET was consistent with strong binding; however, the binding constant could not be determined accurately due to competitive binding with THA+, which leads to a decrease in the Li+@C60˙− absorption upon addition of more than 1 equiv. of 1.
The porphyrin anion-induced electron transfer from TTF-C4P to Li+@C60 did not afford a broad NIR absorption band, which would be expected if the oxidized donor, TTF-C4P˙+, was produced as a free radical cation due to the internal formation of a π-dimer radial cation involving an oxidized TTF˙+ moiety and a neutral TTF subunit likewise present in TTF-C4P˙+.1 The absence of a broad NIR feature thus leads us to suggest that the oxidized host, TTF-C4P˙+, interacts strongly with the reduced Li+@C60˙− guest to produce the supramolecular charge-separated (CS) complex, 1/TTF-C4P˙+/Li+@C60˙−, wherein the porphyrin component remains in its initial, neutral form.
The EPR spectra recorded after electron transfer from TTF-C4P to Li+@C60 in the presence of 1 prior to and during photoirradiation are shown in Fig. 4. Here, the signal at g = 2.0064 is ascribed to TTF-C4P˙+,2,12b,16 whereas the signal at g = 2.0006 is attributed to the Li@C60 radical anion.18
 | ||
| Fig. 4 EPR spectrum of the products of electron transfer from TTF-C4P (0.1 mM) to Li+@C60 (0.1 mM) in the presence of porphyrin 1 (0.1 mM) in PhCN at 298 K after photoirradiation with a high-pressure Hg lamp. | ||
Upon photoirradiation, a new EPR signal at g = 2.0028 appears, which is attributed to the radical cation of porphyrin 1. This finding is consistent with our design expectations for this self-assembled triad, namely that upon photoexcitation of the porphyrin, photoinduced electron transfer takes place from the porphyrin to the TTF-C4P radical cation to form a multi-component, charge-separated complex, 1˙+/TTF-C4P/Li+@C60˙−. This CS state has the charges localized at the two ends of the self-assembled triad. It thus differs dramatically from that obtained in the absence of photoexcitation.
The fate of the triplet excited state (31*) in the supramolecular triad was examined by transient absorption spectroscopy following nanosecond laser flash photolysis measurements. Upon nanosecond laser excitation of a deaerated PhCN solution of 1 (0.1 mM), TTF-C4P (0.1 mM) and Li+@C60 (0.1 mM) at 532 nm, transient absorption bands were observed at 440 nm and 790 nm with bleaching at 650 and 720 nm (Fig. 5a). The absorbance band at 440 nm is attributed to 31*, which decayed with the increase in the absorption bands at 790 nm due to 1˙+ and recovery of bleaching at 650 and 720 nm due to TTF-C4P˙+. Such a spectral change indicates that electron transfer from 31* to TTF-C4P˙+ occurred to produce the higher energy CS state (1˙+/TTF-C4P/Li+@C60˙−). The fact that no changes in the spectral features at 1035 nm ascribable to Li+@C60˙− are observed under conditions of the transient absorption measurements leads us to conclude that the Li+@C60˙− component of the CS state remains the same in the absence and presence of nanosecond laser excitation. On the other hand, the decay of the transient absorption due to 1˙+ is consistent with formation of a higher energy CS state wherein the charges reside on the two termini of the self-assembled triad (i.e., 1˙+/TTF-C4P/Li+@C60˙−). The lifetime of the CS state produced upon photoexcitation is 4.8 μs, as determined from the absorption recovery of the bleaching band at 650 nm ascribed to the porphyrin ground state (Fig. 5). The photodynamics of the supramolecular triad are summarized in the energy diagram given in Scheme 1.
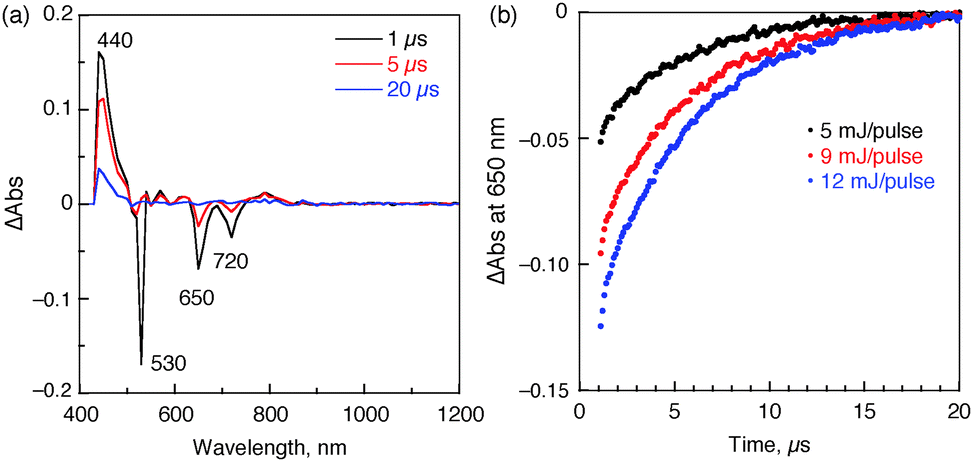 | ||
| Fig. 5 (a) Nanosecond flash photolysis absorption spectra for a mixture of 1 (100 μM), TTF-C4P (100 μM) and Li+@C60 (100 μM) following excitation at 532 nm in deoxygenated PhCN at 298 K as recorded 1 μs (black), 5 μs (red), and 20 μs (blue) after laser pulse irradiation. (b) Decay time profiles of the absorbance at 650 nm obtained using various laser intensities. | ||
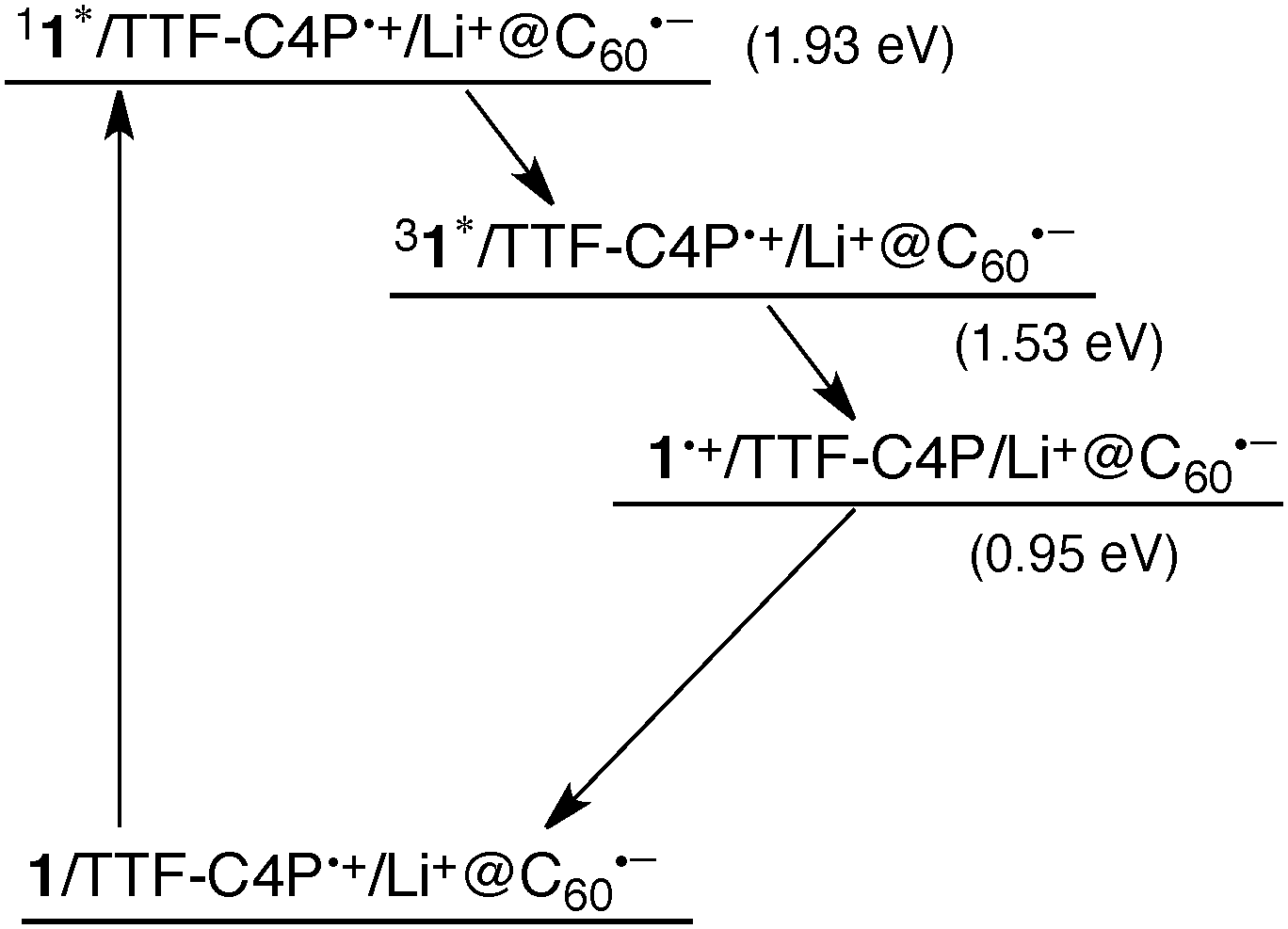 | ||
| Scheme 1 | ||
In conclusion, binding of a porphyrin anion (1) to TTF-C4P results in electron transfer from TTF-C4P to Li+@C60 to produce the supramolecular charge-separated triad (1/TTF-C4P˙+/Li+@C60˙−). Photoexcitation of the porphyrin component (1) within the supramolecular triad leads first to the production of an excited singlet state (11*) and intersystem crossing from 11* to 31*, from which electron transfer to TTF-C4P˙+ occurs to produce the higher energy charge-separated sate (1˙+/TTF-C4P/Li+@C60˙−). The CS lifetime of the latter state was determined to be 4.8 μs. The present study provides a new approach to creating non-covalent, multi-component systems that support the formation of photoinduced charge separated states. Specifically, we detail the construction of supramolecular electron donor–acceptor ensembles via a combination of anion recognition and donor–acceptor interactions and show that it leads to the formation of a well-defined CS state upon photoexcitation. The use of orthogonal recognition motifs is likely to prove to be useful in the preparation of other higher order self assembled ensembles wherein the electronic nature and special arrangement of the individual photo- and redox active components need to be controlled.
This work was supported by Grant-in-Aid (No. 26620154 and 266288037 to K.O.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and ALCA and SENTAN programs from Japan Science Technology Agency (JST) (to S.F.), JSPS Fellows (25627 to Y.K.), Japan, a U.S. NSF grant (CHE 1057904 to J.L.S), and the Robert A. Welch Foundation (grant F-1018 to J.L.S.).
Notes and references
- N. L. Bill, O. Trukhina, J. L. Sessler and T. Torres, Chem. Commun., 2015, 51, 7781 RSC.
- S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938 CrossRef CAS PubMed.
- C. M. Davis, Y. Kawashima, K. Ohkubo, J. M. Lim, D. Kim, S. Fukuzumi and J. L. Sessler, J. Phys. Chem. C, 2014, 118, 13503 CAS.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- (a) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed; (b) F. D. Lewis, R. L. Letsinger and M. R. Wasielewski, Acc. Chem. Res., 2001, 34, 159 CrossRef CAS PubMed.
- (a) M. D. Kaerkaes, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100 CrossRef CAS PubMed; (b) J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS PubMed; (c) J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560 CrossRef CAS PubMed.
- (a) S. Kirner, M. Sekita and D. M. Guldi, Adv. Mater., 2014, 26, 1482 CrossRef CAS PubMed; (b) G. de la Torre, G. Bottari, M. Sekita, A. Hausmann, D. M. Guldi and T. Torres, Chem. Soc. Rev., 2013, 42, 8049 RSC; (c) D. M. Guldi, Phys. Chem. Chem. Phys., 2007, 9, 1400 RSC; (d) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (e) J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53 CrossRef CAS PubMed.
- (a) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609 RSC; (b) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (c) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC; (d) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC; (e) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed.
- (a) F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC; (b) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (c) F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef PubMed; (d) M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30 CrossRef CAS PubMed.
- (a) S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801 RSC; (b) S. Fukuzumi and K. Ohkubo, Dalton Trans., 2013, 42, 1584 Search PubMed.
- (a) Y. Kawashima, K. Ohkubo and S. Fukuzumi, Chem. – Asian J., 2015, 10, 44 CrossRef CAS PubMed; (b) M. Supur, Y. Kawashima, K. R. Larsen, K. Ohkubo, J. O. Jeppesen and S. Fukuzumi, Chem. – Eur. J., 2014, 20, 13976 CrossRef CAS PubMed; (c) M. Supur, Y. Kawashima, Y.-X. Ma, K. Ohkubo, C.-F. Chen and S. Fukuzumi, Chem. Commun., 2014, 50, 15796 RSC.
- (a) Y. Kawashima, K. Ohkubo, K. Mase and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 21166 CrossRef CAS; (b) N. L. Bill, M. Ishida, S. Bahring, J. M. Lim, S. Lee, C. M. Davis, V. M. Lynch, K. A. Nielsen, J. O. Jeppesen, K. Ohkubo, S. Fukuzumi, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 10852 CrossRef CAS PubMed.
- N. L. Bill, M. Ishida, Y. Kawashima, K. Ohkubo, Y. M. Sung, V. M. Lynch, J. M. Lim, D. Kim, J. L. Sessler and S. Fukuzumi, Chem. Sci., 2014, 5, 3888 RSC.
- (a) K. Ohkubo, K. Mase, E. Karnas, J. L. Sessler and S. Fukuzumi, J. Phys. Chem. C, 2014, 118, 18436 CrossRef CAS; (b) J. L. Sessler, E. Karnas, S. K. Kim, Z. Ou, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 15256 CrossRef CAS PubMed.
- For supramolecuar triads and tetrads in which covalently linked dyads and triads are used, see: (a) F. D'Souza, A. N. Amin, M. E. El-Khouly, N. L. Subbaiyan, M. E. Zandler and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 654 CrossRef PubMed; (b) P. Mondal, A. Chaudhary and S. P. Rath, Dalton Trans., 2013, 42, 12381 RSC; (c) V. Bandi, M. E. El-Khouly, K. Ohkubo, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, J. Phys. Chem. C, 2014, 118, 2321 CrossRef CAS.
- J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. Bielawski, T. W. Hudnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324 CrossRef CAS PubMed.
- Y. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2012, 116, 8942 CrossRef CAS PubMed.
- K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic details. See DOI: 10.1039/c5cc03061g |
| This journal is © The Royal Society of Chemistry 2015 |