
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerization of low molecular weight hydrogelators to form electrochromic polymers†
Peter S.
Kubiak
a,
Salmah
Awhida
b,
Christopher
Hotchen
a,
Wentao
Deng
a,
Ben
Alston
b,
Tom O.
McDonald
b,
Dave J.
Adams
*b and
Petra J.
Cameron
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: p.j.cameron@bath.ac.uk
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
First published on 21st May 2015
Abstract
We present a method for the polymerization of low molecular weight hydrogelators to form polymers with unique structures. Carbazole-protected amino acids are shown to form hydrogels by self-assembly into fibrous structures. We show that is possible to directly electropolymerize the hydrogels. This results in the formation of microporous electrochromic polymers with distinctive structure. Polymers formed from the same gelator without the pregelation step show more compact structures. This method opens the possibility of creating polymers templated from pre-assembled gels that have the potential to be used in a wide range of applications.
Poly(carbazoles), poly(fluorenes) and other similar conjugated polymers have great potential as sensors, in light-emitting diodes, and in solar cells.1,2 Many of these polymers exhibit high quantum efficiencies, high thermal stabilities and tunable photoluminescence. They can be synthesized in solution by a range of coupling reactions, or electrochemically where polymerization is initiated by oxidation of the monomer.3 For many of the desired applications, the morphology of the polymers on a surface is important. As a result, there is considerable interest in preparing templated polymers with controlled morphology – for example an increase in porosity or incorporation of a desired structure such as polymer nanowires and nanotubes. As one example of an interesting morphology, conjugated polymer nanowires have many applications.4 Such nanowires often have properties that are different to those of the corresponding thin films or the bulk solids. There are many routes to polymer nanowires, including using hard templates, electrospinning and lithography.4 Another approach is to use the self-assembly of π-conjugated molecules.4,5 Low molecular weight gelators (LMWG), to give one example, self-assemble to form fibrous structures that immobilize the solvent.6
There are many examples of LMWG based on protected amino acids or dipeptides.7–10 Many such LMWG have now been reported where the protecting group on the N-terminus is typically fluorenylmethoxycarbonyl (Fmoc), naphthalene, or another large aromatic group. Gelation can be triggered in water in many ways including a pH change, temperature change, addition of ions, enzyme action, or shear flow. The resulting hydrogels typically contain a network of self-assembled fibers, with the gel properties being highly dependent on the nature of the amino acids in the gelator and how the gelation is carried out.11 There is interest in cross-linking the LMWG to stabilize and strengthen the gel networks, which are often mechanically weak and sensitive to variations in their environment.12 Chemical cross-linking in the gel state has been achieved by photo-crosslinking diacetylene or tyrosine groups on the LMWG.13–15
A number of Fmoc-amino acids can be used as LMWG.16–21 To the best of our knowledge, gels containing Fmoc-amino acids have never been cross-linked, but there is literature precedent for electropolymerising Fmoc-amino acids dissolved in an organic solution (i.e. not in a gel state). For example, Rault-Berthelot et al. grew brittle films of poly(Fmoc-Ala) and poly(Fmoc-Ile) electrochemically from solutions of the protected amino acid monomer in acetonitrile and used them as chiral sensors.22 He et al. electrosynthesised poly(Fmoc-Gly) using boron trifluoride diethyl etherate (BFEE) as the solvent.23 This polymer retained the glycine side chain, was a good blue emitter, and had a conductivity of 0.49 S cm−1. Finally Lai et al. prepared poly(Fmoc-Phe) electrochemically, also using BFEE as a solvent, creating a chiral polymer that was a good blue light emitter.24
In this work, we postulated that the assembly of π-conjugated LMWG into fibers could be used as a pre-structuring step and that polymerization of the fibers could lead to novel microstructures such as conjugated polymer nanowires. To achieve this, we have prepared novel LMWGs based on a range of carbazole amino acids and polymerized the gels in an aqueous environment.25 To the best of our knowledge this is the first example of the electrochemical polymerization of a LMWG. By polymerizing pre-formed gels we have created redox polymers with unique microstructure. We have found that it is possible for π–π stacked gels to undergo a molecular reorganization during polymerization to form conjugated polymers.
As outlined above, we have chosen to electropolymerize gels prepared from carbazole protected amino acids. Carbazole-protected amino acids were chosen as poly(carbazole) is a well-known redox polymer.25,26 In addition, carbazole-protected amino acids were expected to be easier to polymerize compared to Fmoc protected amino acids as polymerization occurs at lower applied voltages (e.g. we found the oxidation onset for Carb-Ala was more than 400 mV less positive than that for Fmoc-Ala in acetonitrile, data not shown). Two carbazole-dipeptide gelators have been reported recently.27 Here, we show that alanine protected at the N-terminus with a carbazole group (Carb-Ala, Scheme 1) is an effective LMWG. A number of other carbazole-protected amino acids form gels and can be polymerized, including valine, glycine and isoleucine, but we focus here on Carb-Ala for clarity. Similarly to other related LMWG (and the reported carbazole-dipeptides27), Carb-Ala can form gels in the bulk via a pH switch in water. At high pH, a free-flowing solution is formed that gels when the pH is lowered, for example on addition of glucono-δ-lactone (GdL, which hydrolyses in situ to gluconic acid28,29). These gels result from the self-assembly of Carb-Ala into fibres to give a transparent gel, with a storage modulus (G′) of 6000 Pa and a loss modulus (G′′) of 700 Pa. The gel was typical for this type of LMWG, with G′ an order of magnitude than G′′ and both being frequency independent.
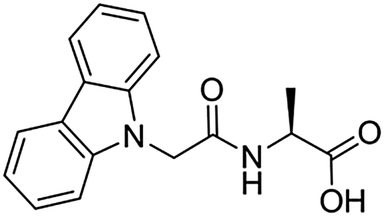 | ||
| Scheme 1 Structure of Carb-Ala. | ||
Spatially resolved, surface supported Carb-Ala gels can also be prepared at an electrode surface via an electrochemically generated pH drop, as we have described previously (Fig. 1a and b).30–32 Briefly, the electrochemical oxidation of hydroquinone to quinone results in a local pH drop at an electrode surface, and hence the gel only forms on the electrode, rather than throughout the system. Again, the gel is a result of the assembly of Carb-Ala into fibres. On drying, an entangled network of fibers can be seen by microscopy, which is again typical for this type of gel (Fig. 1c).
 | ||
| Fig. 1 Photographs of a transparent Carb-Ala gel formed on an FTO-coated glass slide from (a) the top and (b) the side. (c) Shows a SEM image of the xerogel formed by drying the gel in air. | ||
We then attempted to directly polymerize Carb-Ala in the gel phase. A pre-formed gel layer was initially grown on an electrode surface as above. The gel was then removed from the Carb-Ala solution and placed into a 1 mol dm−3 perchloric acid electrolyte (note that there was no free monomer in the bulk solution at this point). The gel was polymerized by repeated cyclic voltammograms (CVs) between 0.2 and 1.1 V (versus Ag/AgCl, 3 mol dm−3). Oxidation of the monomer occurred above 0.8 V. The polymerization process was videoed in real time (video at 16× speed is submitted as part of the ESI†). Fig. 2 shows stills taken from a video of the polymerization of a hemisphere of gel grown on the surface of a 1.6 mm diameter gold disc electrode. At t = 0, there was a transparent hemi-sphere of gel on the electrode surface. As the polymerization progressed, the polymer grew out from the electrode surface and the gel collapsed to become part of the denser polymer film. A small amount of gel can still be seen away from the surface at t = 240 s, but the majority has been incorporated into the polymer. The still at 240 seconds shows the polymer after approximately 8 CVs at 50 mV s−1. Each CV lasted 34 seconds and they were started shortly after t = 0 in the video.
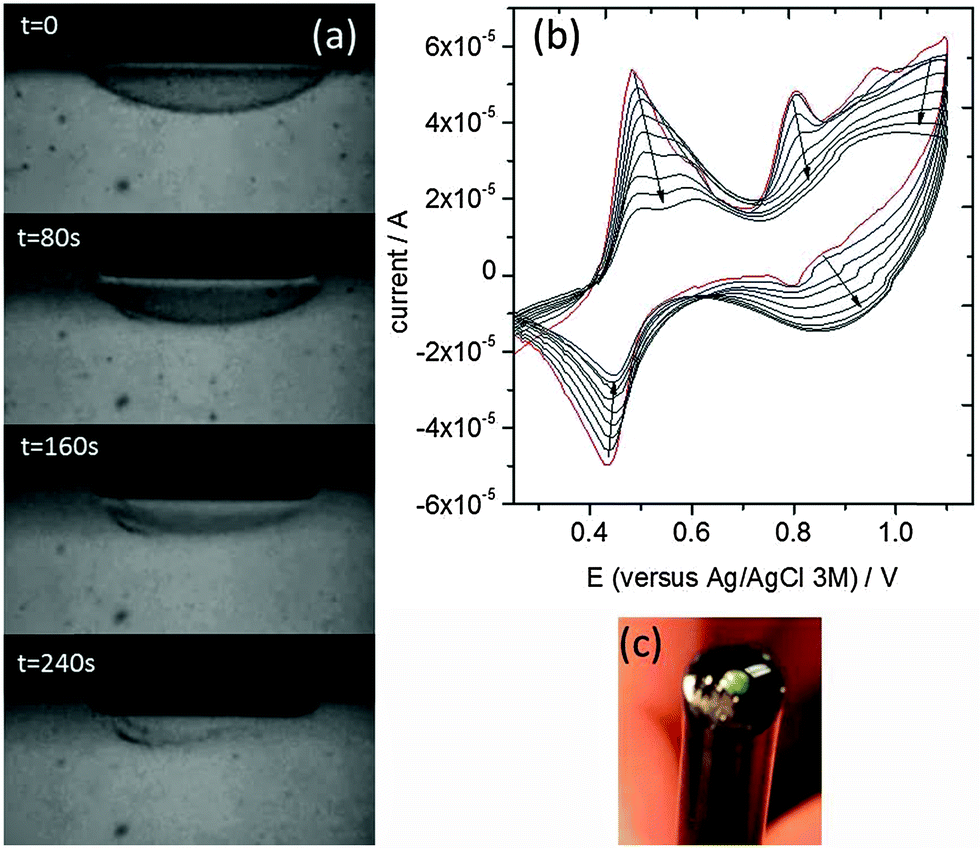 | ||
| Fig. 2 (a) In situ polymerization of a pre formed gel on the surface of a gold disc electrode. At t = 0, the gel is visible as a transparent hemi-sphere above the electrode surface. As the electrode was cycled above the oxidation potential for the monomer, the gel collapsed into a denser polymer layer on the electrode surface. The stills show the progression during the first 8 CVs at 50 mV s−1 (34 seconds per scan). The surface attached polymer was electrochromic and cycled between green and colorless during successive scans. (b) Cyclic voltammograms taken during the polymerization of the gel shown in part (a). The peaks above 0.75 V are connected with the oxidation and reduction of the Carb-Ala monomer/polymer. The arrows show the evolution of the peaks with time. (c) A photograph showing the green polymer grown on the gold disc electrode. | ||
The resulting small area poly(Carb-Ala) films were electrochromic and cycled between green (oxidized form) and colourless (reduced form) with each CV. The CVs taken as a Carb-Ala gel was polymerized are shown in Fig. 2b. The redox process centred on 0.46 V versus Ag/AgCl is due to the hydroquinone/quinone redox couple. As described above, hydroquinone was used in the first step to create a pH drop at the electrode surface and to initiate gelation. The gel was subsequently removed from the hydroquinone solution and placed in 1 mol dm−3 perchloric acid. However, hydroquinone/quinone remains trapped within the gel and shows the quasi-reversible redox peaks centered on 0.46 V. The peaks decrease with increasing numbers of scans, probably due to the collapse of the gel into the polymer and the release of the hydroquinone/quinone back into solution.
The peaks due to monomer and polymer oxidation and reduction occur above 0.75 V and are not as clearly defined as they are for polymerization of drop coated films in the same electrolyte (see Fig. S1, ESI†). However, a broad set of oxidation and re-reduction peaks are observed, centered on approximately 0.92 V. The thin films electropolymerised on 1.6 mm diameter electrodes as shown in Fig. 2 were prepared from gels that were between 0.2 mm to 0.5 mm thick when wet. Thicker films which were several mm thick when wet were also polymerised on FTO slides (see Fig. 3). In addition we have polymerised drop coated films which were much thinner and gave micrometre thick films of polymer (Fig. S5, ESI†). While it is likely that there will be an upper limit to the thickness of the gel layer that can be polymerised, films more than a few mm thick were not investigated in this study.
 | ||
| Fig. 3 A comparison of the structures of large area polymer films grown on FTO glass electrodes. The top row shows the structure of the polymer formed when a gel is electropolymerized. The film grown from the pre-formed gel has a slight brown discoloration due to remaining hydroquinone/quinone within the polymer. The bottom row shows films obtained from the direct electropolymerization of Carb-Ala dissolved in acetonitrile. In both cases, the AFM (films grown from 2 mg mL−1 Carb-Ala) and SEM (films grown from 2.5 mg mL−1 Carb-Ala) images underline the large differences in the microstructures of polymer grown via the gel phase. | ||
As a control, polymers prepared from gels were compared to polymers prepared from Carb-Ala dissolved in acetonitrile (Fig. S2, ESI†). The Carb-Ala does not pre-assemble into fibrous structures in acetonitrile, so polymerization occurs due to oxidation of free monomer in solution. These polymers were also compared to poly(carbazole) prepared by polymerizing carbazole dissolved in acetonitrile (Fig. S3, ESI†). FTIR spectroscopy of the final polymers showed the presence of two peaks at 1660 cm−1 and 1721 cm−1 for the poly(Carb-Ala) films (from LMWGs and from acetonitrile), which were not present in spectra of poly(carbazole) films, showing the retention of the alanine group in the polymer (see Fig. S4, ESI†). Similarly, NMR showed the retention of the alanine group, as well as significant differences compared to the monomer. The spectra were similar for the polymer formed directly from acetonitrile and from the gel film (Fig. S5–S7, ESI†). For both cases, residual monomer can be seen, with the amounts varying depending on whether the polymerization was carried out in acetonitrile or in the gel phase. Due to the polymers only dissolving in DMF and the lack of sufficient difference in refractive index between the polymer and solvent, all attempts at characterizing by GPC failed. Poly(Carb-Ala) produced by polymerization in acetonitrile showed a substantially different structure to poly(carbazole) (Fig. S8, ESI†). Unfortunately, the two systems (polymerisation of a surface bound aqueous gel and or direct polymerisation of the monomer dissolved in acetonitrile) are too different to allow meaningful comparison of the reaction rates.
The structure of the polymers formed via the two-step gelation/electropolymerization process is extremely interesting and quite different to the structure of polymers that did not pass through the templating gel phase. Imaging films where the gel had been polymerized for short times was difficult, as they remained soft and ‘gel-like’ (Fig. S9, ESI† shows a photo of a partially polymerized gel on FTO glass). In order to obtain AFM images for the polymerized gels, the films were left in a covered container overnight to allow them to dry out slightly; it was therefore not possible to see the as-formed structure.
Fig. 3 compares polymers grown from a pre-formed Carb-Ala gel and from Carb-Ala dissolved in acetonitrile. It can be seen that polymers prepared from a pre-formed gel have an open fibrous structure, whereas polymers grown from acetonitrile are much denser. As an additional control, we drop-coated an electrode with Carb-Ala from a THF solution.33 After evaporation of the organic solvent, polymerization of the Carb-Ala was also carried out in 1 mol dm−3 perchloric acid solution. The structure of polymers grown from drop-coated films was quite different to that of polymers grown from pre-formed gels (Fig. S8, ESI† compares the structures of drop coated poly(Carb-Ala) and poly(Carb-Ala) grown from acetonitrile with poly(carbazole) films grown under identical conditions). The drop-coated Carb-Ala polymerized to give needle shaped crystallites growing out from central nucleation points. Our results clearly show that we can use a gel to ‘template’ polymer structure to form unique structures by templating from the fibrous structures formed by the self-assembly of LMWG.
In conclusion, we have shown that Carb-Ala forms fibrous hydrogels in water. The gels can be polymerized by electrochemical oxidation of the carbazole end groups. Polymerized gels gave films with open porous structures, quite different to those prepared when films were grown from Carb-Ala-OH dissolved in MeCN or drop coated onto an electrode and polymerized in perchloric acid. Polymerization of gels was followed in real time; the gel collapsed as the Carb-Ala was incorporated into the polymers. Our data shows that it is possible for π–π stacked gels to undergo a molecular reorganization during polymerization to form conjugated polymers. The polymers were electrochromic (colourless to green) and retained the alanine functionality. These results open the possibility that a wide range of protected peptides could be fully or partially polymerized to form polymers with unique and useful microstructures. Patterning of the electrode could also allow selective polymerization of regions of gel. Overall, this process could lead to the possibility of using partially or completely polymerized hydrogels for a range of sensing and bioelectronics applications – introducing new properties while retaining an open porous structure and the amino acid functionality.
P.J.C. acknowledges the University of Bath and the EPSRC (EP/H026304/1) for funding. S.A. thanks the Libyan government for a studentship. D.J.A. thanks the EPSRC for a Fellowship (EP/L021978/1).
Notes and references
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS PubMed.
- R. C. Advincula, Functional Polymer Films, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, p. 379 Search PubMed.
- J.-F. Morin, M. Leclerc, D. Adès and A. Siove, Macromol. Rapid Commun., 2005, 26, 761 CrossRef PubMed.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2011, 23, 682 CrossRef CAS.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS PubMed.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160 CrossRef CAS PubMed.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150 RSC.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143 RSC.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- S. R. Diegelmann, N. Hartman, N. Markovic and J. D. Tovar, J. Am. Chem. Soc., 2012, 134, 2028 CrossRef CAS PubMed.
- M. de Loos, J. van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675 CrossRef CAS.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2013, 29, 13299 CrossRef CAS PubMed.
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440 CrossRef CAS PubMed.
- D. M. Ryan, S. B. Anderson, F. T. Senguen, R. E. Youngman and B. L. Nilsson, Soft Matter, 2010, 6, 475 RSC.
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Chem. Commun., 2011, 47, 475 RSC.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18 RSC.
- J. Rault-Berthelot, E. Raoult, J. Tahri-Hassani, H. Le Deit and J. Simonet, Electrochim. Acta, 1999, 44, 3409 CrossRef CAS.
- Y. He, W. Guo, M. Pei and G. Zhang, Chin. J. Chem., 2012, 30, 985 CrossRef CAS PubMed.
- C. Lai, W. Guo, X. Tang and M. Pei, Chin. J. Chem., 2012, 30, 507 CrossRef CAS PubMed.
- K. Kawabata and H. Goto, Synth. Met., 2010, 160, 2290 CrossRef CAS PubMed.
- D.-X. Zhuang and P.-Y. Chen, J. Electroanal. Chem., 2009, 626, 197 CrossRef CAS PubMed.
- A. D. Martin, A. B. Robinson and P. Thordarson, J. Mater. Chem. B, 2015, 3, 2277–2280 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- E. K. Johnson, L. Chen, P. S. Kubiak, S. F. McDonald, D. J. Adams and P. J. Cameron, Chem. Commun., 2013, 49, 8698 RSC.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241 RSC.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Am. Chem. Soc., 2010, 132, 5130 CrossRef CAS PubMed.
- G. Inzelt, J. Solid State Electrochem., 2003, 7, 503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for Carb-Ala, further electrochemical and other analysis. See DOI: 10.1039/c5cc03053f |
| This journal is © The Royal Society of Chemistry 2015 |