
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic Cu–amine catalysis for the enantioselective synthesis of chiral cyclohexenones†
A.
Quintard
* and
J.
Rodriguez
*
Aix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France. E-mail: adrien.quintard-inv@univ-amu.fr; jean.rodriguez@univ-amu.fr
First published on 1st May 2015
Abstract
An unprecedented utilization of 1,3-acetonedicarboxylic acid as a 1,3-bis-pro-nucleophile and a reactive acetone surrogate in enantioselective catalysis has been reported. By synergistically activating the ketodiacid by copper catalysis and an α,β-unsaturated aldehyde by amine catalysis, an efficient domino di-decarboxylative Michael/aldol/dehydration sequence takes place leading to valuable chiral cyclohexenones in one single operation in 94 to 99% ee.
To fulfil the ideal of eco-compatible reactions, scientists continuously need to discover innovative activation modes able to perform unprecedented cascade transformations from simple molecules to complex architectures with a perfect control of stereoeselectivity.1 In that regard, synergistic catalysis where different catalytic species are able to selectively activate different reaction partners has recently demonstrated its potential in the discovery of new previously inaccessible chemical transformations notably pioneered by the work from the group of Krische.2 In this particular field, association of copper salts with organocatalysts has proven efficient for the stereoselective activation of numerous nucleophilic and electrophilic partners.3 Notably, copper and iminium activation could be combined synergistically, providing an improved route for the preparation of β-chiral aldehydes.4 1,3-Acetonedicarboxylic acid (1) is an intriguing molecule possessing two pendant carboxylic acid functions that can potentially be involved in double biomimetic-like decarboxylative enolate formation rendering the corresponding acetone α,α′-dianion easily available.5 Prepared on a large scale from raw material namely citric acid, it can act as a potential reactive di-nucleophilic acetone surrogate.6 This property was famously applied by Robinson in his 1917 tropinone synthesis and has since then found intensive utilization in the synthesis of tropinone like skeletons.7 But quite surprisingly, despite its huge potential and to our knowledge there are no examples involving 1 as an acetone α,α′-dianion equivalent in enantioselective catalysis.8 To fill this gap, we initiated a research program to selectively activate this unexploited 1,3-bis-pro-nucleophile in enantioselective synthesis. We first focused on its unaddressed reactivity towards α,β-unsaturated aldehydes as electrophiles in a di-decarboxylative Michael/aldol/dehydration sequence leading to synthetically useful cyclohexenone derivatives, a class of versatile molecules used in natural product synthesis.9 Earlier reports on the preparation of these chiral cyclohexenones required multiple steps such as kinetic resolution of chiral compounds.10 Several groups reported on the organocatalytic condensation of functionalized ketones on α,β-unsaturated aldehydes, leading after cascade cyclization by Knoevenagel or Wittig reaction to chiral cyclohexenones.11 Unfortunately, in all these examples, additional functional groups are present on the obtained cyclohexenone backbones and require subsequent steps for their removal (tert-butyl ester for example). Given the utility of this chiral cyclohexenone motif and its numerous applications, alternative direct access to this structure remains attractive and desirable.
To efficiently apply 1, we initially hypothesized that a secondary amine organocatalyst might activate the electrophile through its iminium ion while a copper salt catalyst, known to promote decarboxylative aldolizations, would activate ketodiacid 1 triggering the overall domino sequence initiated by a decarboxylative Michael addition (Scheme 1).12 Herein we present our results on the development of such biomimetic transformation as well as supplementary successive cascade functionalization of the formed cyclohexenone.
 | ||
| Scheme 1 Proposed cyclohexenone synthesis by synergistic addition of 1 to enals. | ||
We initiated our research by condensing 1 on cinnamaldehyde 2a in the presence of an aminocatalyst able to promote the direct Michael addition via the corresponding iminium intermediate. Selected optimizations are presented in Table 1.13 Among all the solvents tested, MeOH gave the best results. Without any co-catalyst, diaryl prolinol silyl ether cat1 gave the expected product 3a in only 12% yield and a promising 90% ee (entry 1). Gratifyingly and as expected in our proposal, addition of a co-catalyst such as a copper salt considerably increased both yield and enantiocontrol. In the presence of 4 mol% of copper acetate, 3a could be obtained in 37% yield and an excellent 97% ee (entry 2). The increase both in terms of yield and stereocontrol indicates that the copper salt efficiently activates 1, facilitating the enolate formation as well as modifying the transition state of the C–C bond-forming event. Use of other substituents on the diaryl prolinol silyl ether did not improve this result (37–46% yield, 94–96% ee, entries 3–5). Imidazolidinone cat5 also catalyzed the process albeit in a lower 12% yield and 88% ee (entry 6).
|
|
|||
|---|---|---|---|
| Entry | Catalysts | Yieldb (%) | eec (%) |
| a All reactions were run using 1 eq. of 1 (0.2 mmol) and 1.3 eq. of 2 (0.26 mmol) except for entry 16 run on 1 eq. of 1 (1.0 mmol) and 1.6 eq. of 2 (1.6 mmol). b Isolated yield after column chromatography. c Enantiomeric excess determined by chiral GC analysis. | |||
| 1 | cat1 (15%) | 12 | 90 |
| 2 | cat1 (15%), Cu(OAc)2 (4%) | 37 | 97 |
| 3 | cat2 (15%), Cu(OAc)2 (4%) | 37 | 94 |
| 4 | cat3 (15%), Cu(OAc)2 (4%) | 40 | 96 |
| 5 | cat4 (15%), Cu(OAc)2 (4%) | 46 | 94 |
| 6 | cat5 (15%), Cu(OAc)2 (4%) | 12 | 88 |
| 7 | cat1 (15%), PhCOOH (15%) | 24 | 89 |
| 8 | cat1 (15%), imidazole (15%) | 29 | 84 |
| 9 | cat1 (15%), dbu (15%) | 21 | 88 |
| 10 | cat1 (15%), LiOAc (15%) | 18 | 88 |
| 11 | cat1 (15%), LiCl (15%) | 13 | 90 |
| 12 | cat1 (15%), Fe(acac)3 (6%) | 38 | 94 |
| 13 | cat1 (15%), CuI (4%) | 22 | 91 |
| 14 | cat1 (15%), CuTc (4%) | 36 | 95 |
| 15 | cat1 (15%), Cu(i-BuCOO)2 (4%) | 40 | 97 |
| 16 | cat1 (15%), Cu(i-BuCOO)2 (6%) | 50 | >98 |
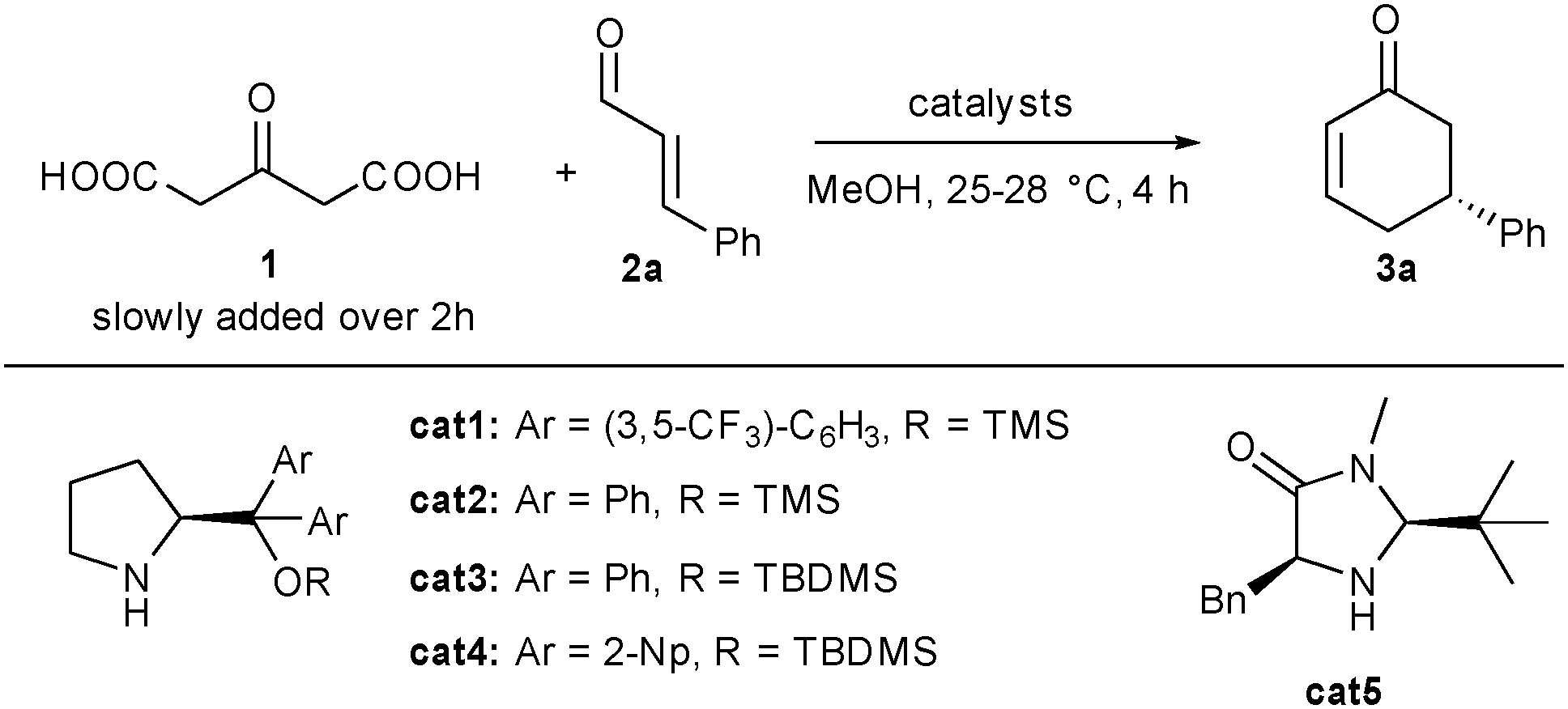
We next turned our attention to the nature of the co-catalyst used to activate the pro-nucleophile 1. Acidic as well as diverse basic additives provided the expected cyclohexenone in lower yields (13 to 29%) as well as decreased enantiocontrol (84 to 90% ee) highlighting the crucial role of the Lewis acid additive (entries 7–11). Other Lewis acids such as iron acetylacetonate and copper iodide or thiophene carboxylate (entries 12–14) were also efficient albeit the results were still lower than with copper acetate (22 to 38% yield and 91–95% ee). Fortunately, turning to copper i-butyrate, both yield and enantiocontrol could be improved (entry 15) and were found to be optimal using 1.6 equivalents of 2a on a 1.6 mmol scale providing 3a in 50% yield and >98% ee (entry 16).
With these optimized conditions in hand, we next investigated the scope of this new dual catalytic domino cycloalkylation. Different α,β-unsaturated aldehydes with diverse substitution patterns could be applied in the transformation (Scheme 2). Electron-withdrawing as well as electron-donating groups could be placed either in the ortho, para or meta position on the aromatic substituent providing the corresponding cyclohexenones 3b–g in 40 to 51% yield and 96 to 99% ee. In the case of electron-withdrawing nitro substituents (3d, 3f and 3g), the temperature had to be decreased to 20 °C to obtain the product probably by limiting both substrate and product decomposition. The aromatic substituent of the aldehyde could be replaced by an ester even though product 3h was obtained with a slightly decreased yield but still a very good enantiocontrol (29% yield and 94% ee). Finally, under the optimized conditions, products starting from aliphatic substituents on the aldehyde, 3i, could not be isolated. Crude NMR showed the formation of the product at around 15–20% together with other unidentified impurities. This indicates that conditions must be further optimized for this family of substrates.
 | ||
| Scheme 2 Scope of the cyclohexenone synthesis by synergistic addition of 1 to enals. | ||
Interestingly, when modulating the conditions by using an excess of ketodiacid 1 in THF, using cat2 and copper acetate, a complementary cascade occurs by addition of a second equivalent of 1 to the enone 3a, directly providing the valuable structure 4a in 87% ee (Scheme 3a, 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). In this transformation, 4a is formed via a multiple cascade process where 4 C–C bonds are destroyed and 3 new C–C and 1 C–H bonds are created. The formation of the terminal CH3 after final addition on the enone is due to the reprotonation of the keto-acid, possibly through another diacid 1 molecule. This reprotonation of the transient enolate is possible in the absence of any other potential electrophile. The enantiomeric excess is the same between 4a and a trace amount of 3a still observed in the reaction mixture. This indicates that 4a forms from cyclohexenone 3a without the intervention of the amino catalyst through copper catalyzed addition.
:1 dr). In this transformation, 4a is formed via a multiple cascade process where 4 C–C bonds are destroyed and 3 new C–C and 1 C–H bonds are created. The formation of the terminal CH3 after final addition on the enone is due to the reprotonation of the keto-acid, possibly through another diacid 1 molecule. This reprotonation of the transient enolate is possible in the absence of any other potential electrophile. The enantiomeric excess is the same between 4a and a trace amount of 3a still observed in the reaction mixture. This indicates that 4a forms from cyclohexenone 3a without the intervention of the amino catalyst through copper catalyzed addition.
 | ||
| Scheme 3 Cascade reaction of 3a and the control experiment. | ||
A mechanistic additional control experiment indicates that the second acid function on 1 is crucial for the cyclization to occur as shown by the partial conversion of ketoacid 5 to the acyclic ketoaldehyde 6 (Scheme 3b).
Mechanistically, the great differences in enantiomeric excesses (98 vs. 90% ee) and yield (50 vs. 12%) observed for cyclohexenone 3a with or without the copper catalyst clearly indicates the presence of a synergistic catalytic mode of action. It is hard to define what accounts for the moderate yields in these reactions since no defined products could be isolated besides 3. The higher enantiomeric excess obtained using copper(II)-i-butyrate as compared to other Lewis acids either arises from a considerable increase in the kinetics of the reaction (limiting the background reaction) or can be due to the ligand ability of isobutyric acid to maximize steric repulsions in the preferred transition state. The fact that higher yields are obtained by slowly adding the ketodiacid 1 to the reacting mixture probably indicates that the copper species formed during the catalytic cycle suffer from a lack of stability (possible reprotonation). Besides these preliminary mechanistic observations, we currently do not know if decarboxylation occurs prior to Michael addition or after the initial C–C bond-forming event. According to work by Shair on copper catalyzed aldolization, C–C bond formation occurs prior to decarboxylation in this process.12h But since in our case we are able to observe decarboxylation in the absence of electrophile (Scheme 3a), it is also possible that using 1, decarboxylation occurs prior to the C–C bond-forming event.14
In conclusion, we have described the first utilization of ketodiacid 1 as a bis-nucleophile and a reactive acetone surrogate in enantioselective catalysis. The key is a synergistic copper/amine dual activation of the ketodiacid and the α,β-unsaturated aldehyde triggering an unprecedented di-decarboxylative Michael/aldol/dehydration domino sequence leading to valuable chiral cyclohexenones in one single operation in 94 to 99% ee. Further mechanistic investigations as well as optimization of aliphatic aldehydes should shed light on the exact reaction mechanism and would probably improve the reaction scope. We are convinced that this study will open the way for multiple applications of this particularly interesting substrate as well as the dual copper–iminium activation.
The Agence Nationale pour la Recherche (ANR-13-PDOC-0007-01), the Centre National de la Recherche Scientifique (CNRS) and the Aix-Marseille Université are gratefully acknowledged for financial support. The authors warmly thank Marion Jean and Nicolas Vanthuyne (Aix-Marseille Université) for chiral-phase HPLC analysis.
Notes and references
- For reviews on economies in synthesis, see: (a) B. M. Trost, Science, 1991, 254, 1471 CAS; (b) P. A. Wender, F. C. Bi, G. G. Gamber, F. Gosselin, R. D. Hubbard, M. J. C. Scanio, R. Sun and T. J. Williams, Pure Appl. Chem., 2002, 74, 25 CrossRef CAS; (c) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed . For selected reviews on cascade and organocascade reactions, see: ; (d) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed; (e) K. C. Nicolaou and K. J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993 RSC; (f) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (g) D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Chem. – Eur. J., 2013, 19, 2218 CrossRef CAS PubMed; (h) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- For the pioneering combination of metal and organocatalysis, see: (a) B. G. Jellerichs, J.-R. Kong and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 7758 CrossRef CAS PubMed . For a selected review on the topic, see: ; (b) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- Combination of tertiary amines catalysis with copper catalysis: (a) K. R. Knudsen and K. A. Jørgensen, Org. Biomol. Chem., 2005, 3, 1362 RSC; (b) T. Yang, A. Ferrali, F. Sladojevich, L. Campbell and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 9140 CrossRef CAS PubMed; (c) C.-L. Ren, T. Zhang, X.-Y. Wang, T. Wu, J. Ma, Q.-Q. Xuan, F. Wei, H.-Y. Huang, D. Wang and L. Liu, Org. Biomol. Chem., 2014, 12, 9881 RSC . Combination of hydrogen bonding catalysis with copper catalysis: ; (d) Y. Lu, T. C. Johnstone and B. A. Arndtsen, J. Am. Chem. Soc., 2009, 131, 11284 CrossRef CAS PubMed ; Combination of enamine activation with copper catalysis: ; (e) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986 CrossRef CAS PubMed; (f) A. Quintard and A. Alexakis, Adv. Synth. Catal., 2010, 352, 1856 CrossRef CAS PubMed; (g) A. Yoshida, M. Ikeda, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 592 CrossRef CAS PubMed; (h) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS PubMed; (i) S. P. Simonovich, J. F. Van Humbeck and D. W. C. MacMillan, Chem. Sci., 2012, 3, 58 RSC.
- For the combination of iminium activation and copper catalysis, see: (a) S. Afewerki, P. Breistein, K. Pirttila, L. Deiana, P. Dziedzic, I. Ibrahem and A. Córdova, Chem. – Eur. J., 2011, 17, 8784 CrossRef CAS PubMed; (b) I. Ibrahem, S. Santoro, F. Himo and A. Córdova, Adv. Synth. Catal., 2011, 353, 245 CrossRef CAS PubMed; (c) I. Ibrahem, P. Breistein and A. Córdova, Angew. Chem., Int. Ed., 2011, 50, 12036 CrossRef CAS PubMed; (d) Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756 CrossRef CAS PubMed . For other examples of Lewis-acid iminium catalysis: ; (e) M. Meazza, V. Ceban, M. B. Pitak, S. J. Coles and R. Rios, Chem. – Eur. J., 2014, 20, 16853 CrossRef CAS PubMed; (f) V. Ceban, P. Putaj, M. Meazza, M. B. Pitak, S. J. Coles, J. Vesely and R. Rios, Chem. Commun., 2014, 50, 7447 RSC.
- For reviews on catalytic decarboxylative additions, see: (a) Y. Pan and C.-H. Tan, Synthesis, 2011, 2044 CAS; (b) Z.-L. Wang, Adv. Synth. Catal., 2013, 355, 2745 CrossRef CAS PubMed; (c) S. Nakamura, Org. Biomol. Chem., 2014, 12, 394 RSC.
- R. Adams, H. M. Chiles and C. F. Rassweiler, Org. Synth., 1925, 5, 5 CrossRef.
- Robinson used the calcium salt of 1 as a reactive pro-nucleophile: R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762 RSC.
- For interesting limited examples of applications of 1 in non-stereoselective catalyzed reactions see: (a) C. Li and B. Breit, J. Am. Chem. Soc., 2014, 136, 862 CrossRef CAS PubMed; (b) B. E. Boughton, M. D. W. Griffin, P. A. O'Donnell, R. C. J. Dobson, M. A. Perugini, M. A. Gerrard and C. A. Hutton, Bioorg. Med. Chem., 2008, 16, 9975 CrossRef CAS PubMed.
- For examples of synthetic applications of these cyclohexenones, see: (a) H. M. Ge, L.-D. Zhang, R. X. Tan and Z.-J. Yao, J. Am. Chem. Soc., 2012, 134, 12323 CrossRef CAS PubMed; (b) X.-M. Zhang, H. Shao, Y.-Q. Tu, F.-M. Zhang and S.-H. Wang, J. Org. Chem., 2012, 77, 8174 CrossRef CAS PubMed; (c) K. Xu, B. Cheng, Y. Li, T. Xu, C. Yu, J. Zhang, Z. Ma and H. Zhai, Org. Lett., 2014, 16, 196 CrossRef CAS PubMed.
- (a) M. Asaoka, K. Shima, N. Fujii and H. Takei, Tetrahedron, 1988, 44, 4757 CrossRef CAS; (b) M. Asaoka, K. Shima and H. Takei, J. Chem. Soc., Chem. Commun., 1988, 430 RSC; (c) M. Asaoka, K. Nishimura and H. Takei, Bull. Chem. Soc. Jpn., 1990, 63, 407 CrossRef CAS; (d) R. Naasz, L. A. Arnold, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 927 CrossRef CAS; (e) Q. Chen, M. Kuriyama, T. Soeta, X. Hao, K.-I. Yamada and K. Tomioka, Org. Lett., 2005, 7, 4439 CrossRef CAS PubMed; (f) M. S. Taylor, D. N. Zalatan, A. M. Lerchner and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 1313 CrossRef CAS PubMed; (g) T. Soeta, K. Selim, M. Kuriyama and K. Tomioka, Tetrahedron, 2007, 63, 6573 CrossRef CAS PubMed; (h) R. D. Carpenter, J. C. Fettinger, K. S. Lam and M. J. Kurth, Angew. Chem., Int. Ed., 2008, 47, 6407 CrossRef CAS PubMed; (i) A. Kolb, S. Hirner, K. Harms and P. von Zezschwitz, Org. Lett., 2012, 14, 1978 CrossRef CAS PubMed.
- For a review on organocatalytic construction of cyclohexenones, see: (a) X. Yang, J. Wang and P. Li, Org. Biomol. Chem., 2014, 12, 2499 RSC . For a review on organocatalytic cyclization: ; (b) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed . For the two steps synthesis of cyclohexenones of type of 3, see: ; (c) A. Carlone, M. Marigo, C. North, A. Landa and A. K. A. Jørgensen, Chem. Commun., 2006, 4928 RSC . For selected examples of other organocatalytic cyclizations leading to cyclohexenones with divers functionalities: ; (d) M. Marigo, S. Bertelsen, A. Landa and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 5475 CrossRef CAS PubMed; (e) P. Bolze, G. Dickmeiss and K. A. Jørgensen, Org. Lett., 2008, 10, 3753 CrossRef CAS PubMed; (f) Y. Hayashi, M. Toyoshima, H. Gotoh and H. Ishikawa, Org. Lett., 2009, 11, 45 CrossRef CAS PubMed; (g) Y.-K. Liu, C. Ma, K. Jiang, T.-Y. Liu and Y.-C. Chen, Org. Lett., 2009, 11, 2848 CrossRef CAS PubMed; (h) L. Albrecht, B. Richter, C. Vila, H. Krawczyk and K. A. Jørgensen, Chem. – Eur. J., 2009, 15, 3093 CrossRef CAS PubMed.
- For a previous decarboxylative Michael addition on α,β-unsaturated aldehydes, see: (a) Q. Ren, S. Sun, J. Huang, W. Li, M. Wu, H. Guo and J. Wang, Chem. Commun., 2014, 50, 6137 RSC . For selected examples of nickel or organocatalyzed decarboxylative 1,4-additions: ; (b) D. A. Evans, S. Mito and S. Seidel, J. Am. Chem. Soc., 2007, 129, 11583 CrossRef CAS PubMed; (c) J. Lubkoll and H. Wennemers, Angew. Chem., Int. Ed., 2007, 46, 6841 CrossRef CAS PubMed; (d) H. W. Moon and D. Y. Kim, Tetrahedron Lett., 2012, 53, 6569 CrossRef CAS PubMed; (e) Y. K. Kang, H. J. Lee, H. W. Moon and D. Y. Kim, RSC Adv., 2013, 3, 1332 RSC . For examples of copper catalyzed decarboxylative aldolizations: ; (f) G. Lalic, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2003, 125, 2852 CrossRef CAS PubMed; (g) D. Magdziak, G. Lalic, H. M. Lee, K. C. Fortner, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2005, 127, 7284 CrossRef CAS PubMed; (h) K. C. Fortner and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 1032 CrossRef CAS PubMed; (i) L. Yin, M. Kanai and M. Shibasaki, Tetrahedron, 2012, 68, 3497 CrossRef CAS PubMed.
- See ESI† for additional details on the reaction optimization.
- See ESI† for mechanistic hypothesis on the catalytic cycle.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, optimization tables, and experimental and spectroscopic data for all compounds. See DOI: 10.1039/c5cc02987b |
| This journal is © The Royal Society of Chemistry 2015 |