
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
KOtBu-mediated annulation of acetonitrile with aldehyde: synthesis of substituted dihydropyridin-2(1H)-ones, pyridin-2(1H)-ones, and thiopyridin-2(1H)-ones†
Abhimanyu
Yadav
,
Ajay
Verma
 ,
Saket
Patel
,
Amit
Kumar
,
Vandana
Rathore
,
Meenakshi
,
Shailesh
Kumar
and
Sangit
Kumar
*
,
Saket
Patel
,
Amit
Kumar
,
Vandana
Rathore
,
Meenakshi
,
Shailesh
Kumar
and
Sangit
Kumar
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, Indore By-pass Road, Bhauri, Bhopal, Madhya Pradesh 462 066, India. E-mail: sangitkumar@iiserb.ac.in
First published on 4th June 2015
Abstract
The KOtBu-mediated annulation of acetonitrile with aldehyde was observed, in which the cleavage of four C(sp3)–H bonds occurred and a total of eight new bonds were formed during the synthesis of substituted dihydropyridinones in the presence of peroxide. Furthermore, dihydropyridinones have been transformed into pyridinones using KOtBu in DMSO.
Annulation by the condensation of readily available substrates, in which the formation of several new bonds occurred by the coupling of C–H bonds in a single pot, is seen as an attractive approach for the preparation of heterocycles.1 This strategy avoids prefunctionalized coupling partners, particularly, halogenated substrates, which generates H2O as waste, and expands the substrate scope.
Heterocycles, particularly, N-containing, such as dihydropyridin-2(1H)-ones, pyridin-2(1H)-ones, and substituted pyridines, are privileged structures with various biological and medicinal properties.2,3 Dihydropyridin-2(1H)-one analogues are being used as hypertensive drugs for calcium channel blockage and for the treatment of diabetes, obesity, and neuropeptide.3 In addition, dihydropyridin-2(1H)-one core is present in the natural products such as homoclausenamide, batzelladine, and carambine alkaloids, which possess HIV-gp-120CD4 inhibition and selective α1a receptor antagonist activities.4
In view of their biological importance, several synthetic methods have been presented in the literature (eqn (1), Scheme 1).5–13 The coupling of α,β-unsaturated acid chloride and ester with enaminonitrile and sodium cyanomethanide, respectively, have been studied for the synthesis of dihydropyridinones.5–8,11,12 Bode et al. and Biju et al. synthesized dihydropyridinones enantioselectively by the coupling of α,β-unsaturated aldehydes with various imine derivatives catalysed by N-heterocyclic carbene.10 Nonetheless, these methods required prefunctionalized substrates to obtain dihydropyridin-2(1H)-ones and pyridin-2(1H)-ones. Prefunctionalized substrates, such as α,β-unsaturated acyl chloride, esters, sodium cyanomethanide, and β-aminonitrile, are either expensive or difficult to handle. The use of prefunctionalized substrates in the coupling reactions restricts the substrate scope because of the difficulty in their synthesis and also due to their incompatibility with another coupling partner.
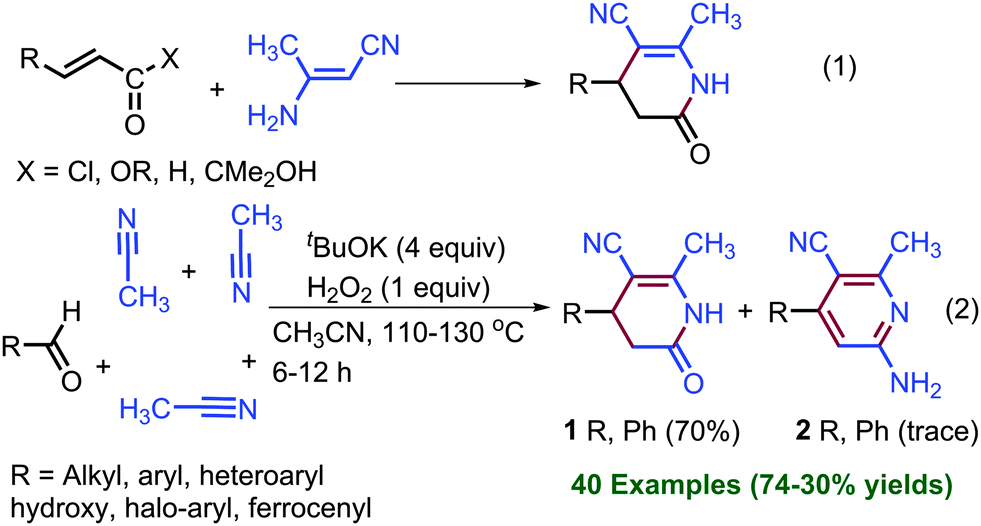 | ||
| Scheme 1 Synthesis of dihydropyridin-2(1H)-ones. | ||
TM-free KOtBu-mediated C–C and C–X coupling reactions between C–H and C–X (X, halogens) bonds and cross coupling between two C–H bonds have been studied by us and others.14–17 Herein, we present a KOtBu base-mediated coupling reaction between aldehyde and acetonitrile for the synthesis of dihydropyridin-2(1H)-ones without employing prefunctionalized substrates (eqn (2)). In this coupling reaction, the cleavage of four sp3-C–H bonds was observed, and total eight new bonds formed. Furthermore, synthesized dihydropyridin-2(1H)-ones have been oxidized into pyridin-2(1H)-ones using the novel approach of KOtBu in DMSO.
After the screening of various bases and additives for the coupling of acetonitrile with the aldehyde at 110 °C in a sealed tube (see ESI,† pages S2–S4, for optimization), we chose one mmol of aldehyde, four mmol of KOtBu base and one mmol of aq. H2O2 in excess of CH3CN (four mL) for the preparation of dihydropyridin-2(1H)-ones. The results are summarized in Scheme 2. Dihydropyridin-2(1H)-one 1 was obtained in 70% yield by the condensation of benzaldehyde with acetonitrile.
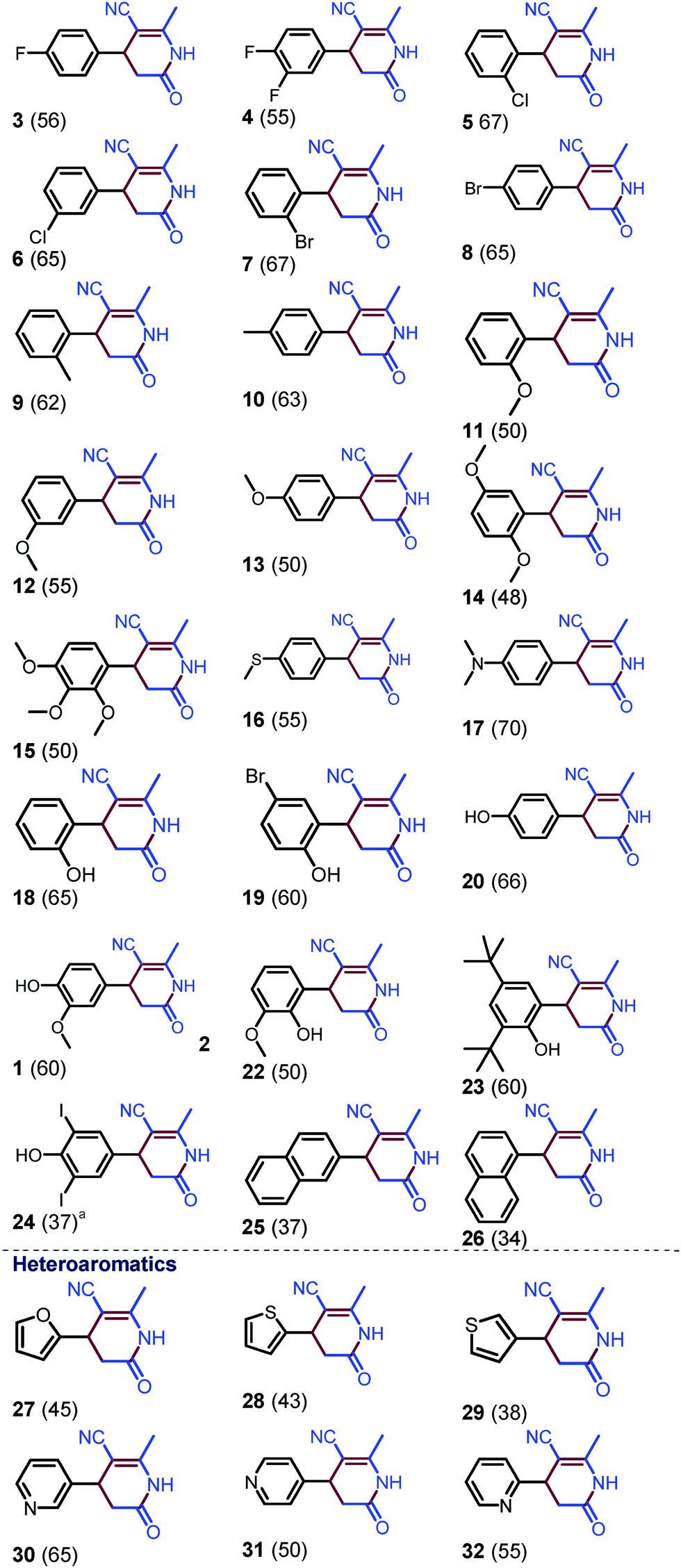 | ||
| Scheme 2 Substrate scope with regard to aromatic aldehydes. Reaction was carried by heating aldehyde (1 mmol), KOtBu (4 mmol), and H2O2 (1 mmol, 30% aq. solution), and CH3CN (4 mL) in a sealed tube at 110 °C. Yields were reported with respect to aryl aldehyde. a Heated at 130 °C. | ||
The formation of 4-phenyl-pyridine 2 was also observed as a minor product and could only be confirmed by mass analysis (see ESI,† page S3). After the synthesis of 1, halogen-substituted benzaldehydes were subjected to the coupling reaction. Fluoro, difluoro, chloro, and bromo substituted benzaldehydes are well tolerated under the optimized reaction conditions and halogen-substituted dihydropyridin-2(1H)-ones 3–7 were obtained in 55–67% yields. Electron-donating substituents, such as CH3, mono, di, and tri-OCH3, SCH3, and N(CH3)2 on benzaldehyde, have also shown compatibility under the reaction conditions and produced respective dihydropyridin-2(1H)-ones 9–17 in 48–70% yields. Interestingly, benzaldehyde-containing acidic OH protons also reacted with acetonitrile and formed hydroxy substituted dihydropyridin-2(1H)-ones 18–24. Various other aromatic aldehydes, such as naphthyl, furanyl, thiophenyl, and pyridyl aldehydes, also underwent coupling reactions with acetonitrile to provide naphthyl and heteroaryl dihydropyridin-2(1H)-ones 25–32 in 34–65% yields.
Alkyl aldehydes were then subjected to the coupling reaction with acetonitrile (Scheme 3). Indeed, alkyl aldehydes provided 4-alkyl substituted dihydropyridin-2(1H)-ones, such as methyl, n-butyl, n-decyl, iso-propyl, and cyclohexyl dihydropyridin-2(1H)-ones 33–40, in moderate yields, although a high temperature (130 °C) is required to accomplish the annulation (please see ESI,† page S18).
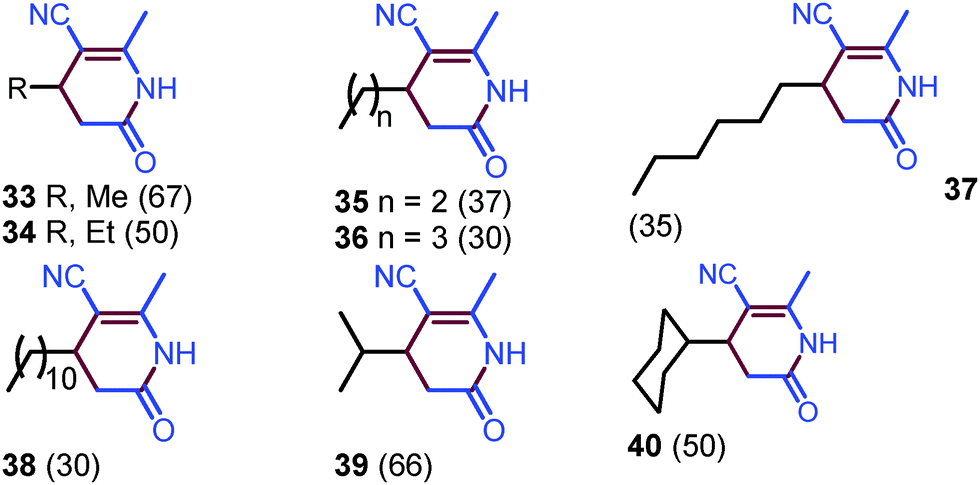 | ||
| Scheme 3 Substrate scope with regards to alkyl aldehydes. | ||
Synthesized dihydro- and pyridin-2(1H)-ones 1, 5, 16 and 42 were also characterized by single crystal X-ray structural study (Fig. 1; for details, see ESI,† pages S32–S77).18
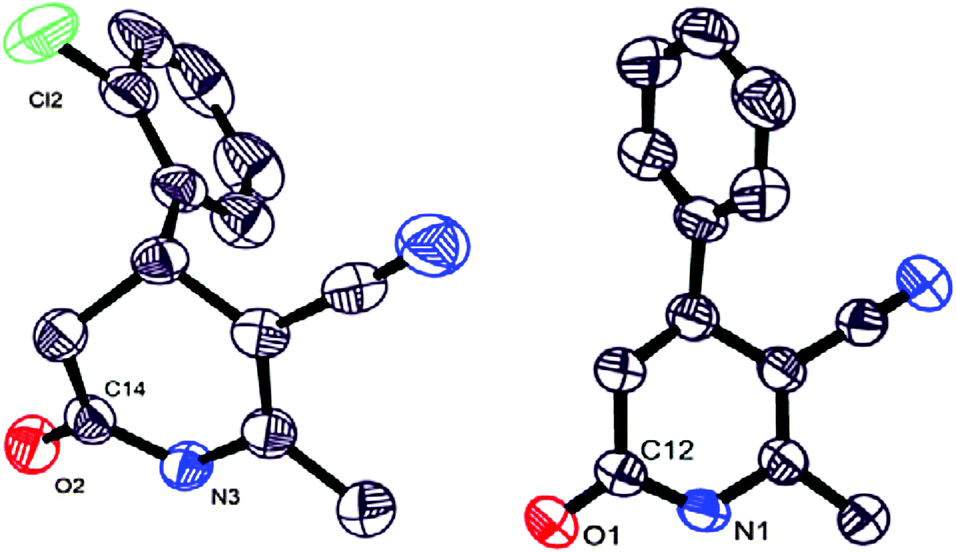 | ||
| Fig. 1 Crystalline structures of dihydro- and pyridin-2(1H)-ones 5 and 42. | ||
Under optimized conditions, when ferrocene aldehyde was subjected for coupling with acetonitrile, 4-ferrocene substituted pyridine 41 was obtained in 76% yield instead of the expected ferrocenyl dihydropyridin-2(1H)-one (Scheme 4). The structure of 41 is established by single-crystal X-ray study.
 | ||
| Scheme 4 Synthesis of 4-ferrocenyl pyridine under optimized conditions. | ||
Further utilities of synthesized dihydropyridin-2(1H)-ones were explored (Scheme 5). The addition of KOtBu to dihydropyridin-2(1H)-ones in DMSO provided selective oxidation of C–H bonds leading to pyridin-2(1H)-ones 42–47 in 35–80% yields. It appears that DMSO not only acts as a solvent but also as an oxidizing agent. The oxidation of dihydropyridin-2(1H)-ones into pyridin-2(1H)-ones could not be achieved in acetonitrile even in the presence of excess KOtBu and oxidant H2O2, and an excess of KOtBu led to the self-coupling of acetonitrile (see ESI,† page S29).
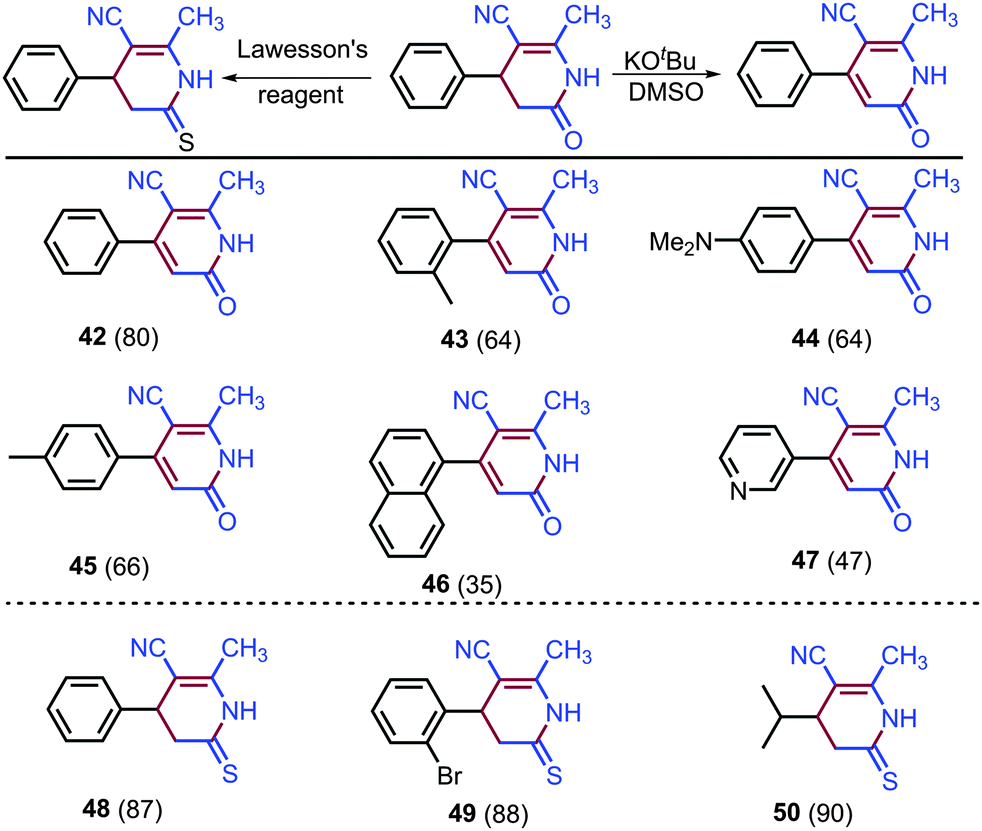 | ||
| Scheme 5 Conversion of dihydropyridin-2(1H)-ones into pyridin-2(1H)-ones and thio analogues. | ||
Synthesized 3,4-dihydropyridin-2(1H)-ones were then transformed into dihydropyridine-2(1H)-thiones (Scheme 5), which exhibit vasodilator, cardiotonic, and antitumor biological activities and also show enriched coordination chemistry as ligands.8,19 The addition of Lawesson's reagent to dihydropyridin-2(1H)-ones gave respective thio analogues 48–50 in 87–90% yields.
Deuterated acetonitrile was then made to react with the benzaldehyde to gain mechanistic insight (Scheme 6). The obtained heterocycle 51 shows the incorporation of five deuterium atoms as studied by mass spectrometry. A deuterium–hydrogen exchange at the first and sixth positions of 3 was also observed and could be rationalized by mechanistic understanding (vide infra). When reaction was performed on (E/Z)-but-2-enal substrate under optimized conditions, dihydropyridin-2(1H)-one 33 was obtained, which was also formed by the reaction of acetaldehyde with acetonitrile (Scheme 3, vide supra).
 | ||
| Scheme 6 Control reactions with CD3CN and with 2-alkene aldehyde. | ||
In the tentative mechanism (Scheme 7), the deprotonation of acetonitrile in the presence of KOtBu would lead to cyanomethanide, which adds to the second molecule of CH3CN, forming (1-cyanopropan-2-ylidene)amide I.21 This may form carbanion IIvia proton transfer, which then adds to aldehyde followed by H2O removal, providing (3-cyanobut-3-en-2-ylidene)amide III. This may react with the third CH3CN molecule to generate intermediate IV, which may convert into carbanion V by a 1,3-proton transfer. An intramolecular attack of carbanion V to the benzylic carbon would furnish the cyclized carbanion VI, which would undergo resonance, forming VII. Intermediate VII may be hydrolyzed to yield 3,4-dihydropyridin-2(1H)-one 1. Although the exact role of H2O2 is not known in the reaction, the formation of ammonia and the improved yield of 1 was observed in the presence of H2O2. This suggests that H2O2 facilitates NH hydrolysis into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.
O group.
 | ||
| Scheme 7 Proposed mechanism for the coupling of CH3CN.20 | ||
In summary, we have shown that substituted dihydropyridin-2(1H)-ones can be synthesized from simple aldehyde and acetonitrile by employing KOtBu base without the use of prefunctionalized substrates. A wide range of aldehydes, including aliphatic; diversely substituted aromatics, including naphthalenes; and heteroaromatics, such as thiophenes, pyridines, and furans; coupled with the three molecules of acetonitrile. Moreover, dihydropyridin-2(1H)-ones have been oxidized into pyridin-2(1H)-ones through a novel method using KOtBu in DMSO. We are currently exploring the scope of the coupling reaction, particularly, for the synthesis of substituted pyridines from readily available substrates utilizing the KOtBu base.
We thank IISER Bhopal, DST and DRDO, New Delhi, for generous funding. AY, AV, VR thank CSIR-UGC, New Delhi; AK, acknowledges CSIR, New Delhi; and SP and Meenakshi grateful to DRDO for fellowships.
Notes and references
- J. Xie, H. Jiang, Y. Cheng and C. Zhu, Chem. Commun., 2012, 48, 979 RSC; R. Samanta, J. Lategahn and A. P. Antonchick, Chem. Commun., 2012, 48, 3194 RSC; M. Potowski, A. P. Antonchick and H. Waldmann, Chem. Commun., 2013, 49, 7800 RSC; K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS PubMed; T. Kaicharla, M. Thangaraj and A. T. Biju, Org. Lett., 2014, 16, 1728 CrossRef PubMed; S. R. Yetra, T. Roy, A. Bhunia, D. Porwal and A. T. Biju, J. Org. Chem., 2014, 79, 4245 CrossRef PubMed; S. Manna, R. Narayan, C. Golz, C. Strohmann and A. P. Antonchick, Chem. Commun., 2015, 51, 6119 RSC . Construction of pyridine and isoquinoline: K. Wu, Z. Huang, C. Liu, H. Zhang and A. Lei, Chem. Commun., 2015, 51, 2286 RSC; S. Manna, R. Narayan, C. Golz, C. Strohmann and A. P. Antonchick, Chem. Commun., 2015, 51, 6119 RSC; S. R. Yetra, S. Mondal, E. Suresh and A. T. Biju, Org. Lett., 2015, 17, 1417 CrossRef PubMed; C. Pan, H. Zhang, J. Han, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 3786 RSC; H. Zhang, C. Pan, N. Jin, Z. Gu, H. Hu and C. Zhu, Chem. Commun., 2015, 51, 1320 RSC.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- Anti-inflammatory, antibacterial and antifungal agent: A. Bhatewara, S. R. Jetti, T. Kadre, P. Paliwal and S. Jain, Int. J. Med. Chem., 2013, 197612 Search PubMed . Antagonist: P. G. Nantermet, J. C. Barrow, H. G. Selnick, C. F. Homnick, R. M. Freidinger, R. S. L. Chang, S. S. O'Malley, D. R. Reiss, T. P. Broten, R. W. Ransom, D. J. Pettibone, T. Olahc and C. Forray, Bioorg. Med. Chem. Lett., 2000, 10, 1625 CrossRef CAS . Rho-Kinase inhibitor: K. B. Goodman, H. Cui, S. E. Dowdell, D. E. Gaitanopoulos, R. L. Ivy, C. A. Sehon, R. A. Stavenger, G. Z. Wang, A. Q. Viet, W. Xu, G. Ye, S. F. Semus, C. Evans, H. E. Fries, L. J. Jolivette, R. B. Kirkpatrick, E. Dul, S. S. Khandekar, T. Yi, D. K. Jung, L. L. Wright, G. K. Smith, D. J. Behm, R. Bentley, C. R. P. Doe, E. Hu and D. Lee, J. Med. Chem., 2007, 50, 6 CrossRef PubMed.
- M.-H. Yang, Y.-Y. Chen and L. Huang, Phytochemistry, 1988, 27, 445 CrossRef CAS.
- A. Uchida, A. Doyama and S. Matsuda, Bull. Chem. Soc. Jpn., 1970, 42, 963 CrossRef.
- S. K. D. Chowdhury, M. Sarkar, S. R. Chowdhury and K. K. Mahalanabis, Synth. Commun., 1996, 26, 4233 CrossRef PubMed.
- By Hantzsch reaction: S. Y. Torres, E. Ochoa, Y. Verdecia and F. Rebolledo, Tetrahedron, 2014, 70, 4675 CrossRef CAS PubMed.
- For conversion of pyridinones to thiones: S. K. D. Chowdhury, M. Sarkar, A. Chatterjee and K. K. Mahalanabis, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2003, 42, 2563 Search PubMed.
- By ring transformation: M. M. Marugán, N. Martín, C. Seoanc and J. L. Soto, Liebigs Ann. Chem., 1989, 145 CrossRef PubMed.
- NHC catalyzed: B. Wanner, J. Mahatthananchai and J. W. Bode, Org. Lett., 2011, 13, 5378 CrossRef CAS PubMed . Coupling of 2-bromoenals with enamines: S. R. Yetra, A. Bhunia, A. Patra, M. V. Mane, K. Vanka and A. T. Biju, Adv. Synth. Catal., 2013, 355, 1089 CrossRef PubMed.
- W. Goehring, P. J. Harrington, L. M. Hodges, D. A. Johnston and G. Rimmler, US Pat., 20050014792A1, 2005 Search PubMed.
- H. Turdi, J. J. Rangeland, R. M. Lawrence, D. Cheng, S. Ahmad, W. Meng, R. P. Brigance and P. Devasthale, US Pat., 20130143843A1, 2013 Search PubMed.
- H. G. Haertwig, V. M. Li, U. Rosentreter, K.-H. Schlemmer, S. Allerheiligen, L. Telan, L. Bärfacker, J. Keldenich, M. F. Fitzgerald, K. Nash, B. Albrecht and D. Meurer, US Pat., 007230017B2, 2007 Search PubMed.
- Our recent work on KOtBu-mediated C–C and C–X bond forming reactions: (a) For C–X Coupling: A. Kumar, B. S. Bhakuni, C. D. Prasad, S. Kumar and S. Kumar, Tetrahedron, 2013, 69, 6383 Search PubMed; (b) A. Kumar, A. Yadav, A. Verma, S. Jana, M. Sattar, S. Kumar, C. D. Prasad and S. Kumar, Chem. Commun., 2014, 50, 9481 RSC; (c) S. Kumar, V. Rathore, A. Verma, C. D. Prasad, A. Kumar, A. Yadav, S. Jana, M. Sattar, Meenakshi and S. Kumar, Org. Lett., 2015, 17, 82 CrossRef CAS PubMed.
- KOtBu-mediated coupling reactions: S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673 Search PubMed; C.-L. Sun, H. Li, D.-G. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zhang, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 Search PubMed; C.-L. Sun, Y.-F. Gu, B. Wang and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 10844 Search PubMed; C.-L. Sun, Y.-F. Gu, W.-P. Huang and Z.-J. Shi, Chem. Commun., 2011, 47, 9813 Search PubMed.
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 Search PubMed; H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 12542 Search PubMed; E. Doni, S. Zhou and J. A. Murphy, Molecules, 2015, 20, 1755 Search PubMed; S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818 Search PubMed; S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476 Search PubMed; H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545 Search PubMed.
- Y. Wu, P. Y. Choy and F. Y. Kwong, Asian J. Org. Chem., 2014, 3, 1262 Search PubMed; T. L. Chan, Y. Wu, P. Y. Choy and F. Y. Kwong, Chem. – Eur. J., 2013, 19, 15802 Search PubMed; Y. Wu, P. Y. Choy and F. Y. Kwong, Org. Biomol. Chem., 2014, 12, 6820 Search PubMed.
- Single crystals were obtained by slow evaporation of CH2Cl2 solution, 5 crystallized in chiral P21 space group.
- P. D. Arkivos, Coord. Chem. Rev., 2001, 213, 181 Search PubMed.
- On the suggestion of one of the reviewer, reaction mixture of PhCHO (1 equiv.), CH3CN, KOtBu (4 equiv.), and H2O2 (1 equiv.) was monitored by EPR spectroscopy. The formation of radical species could not be confirmed by EPR in the reaction mixture which suggests that reaction may not proceed via radical pathway.
- (1-Cyanopropan-2-ylidene)amide I has been prepared earlier by the treatment of sodium with acetonitrile, ref. 5. It is unlikely that reaction involved radicals. For coupling of acetonirle via radical˙ CH2CN pathway: C. Pan, H. Zhang and C. Zhu, Org. Biomol. Chem., 2015, 13, 361 Search PubMed; A. Bunescu, Q. Wang and J. Zhu, Chem. – Eur. J., 2014, 20, 14633 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectra, crystal data. CCDC 1038679 (1), 1038680 (5), 1057417 (16), 1057418 (41), 1038678 (42). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02964c |
| This journal is © The Royal Society of Chemistry 2015 |