
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-step solid-state synthesis of PEPPSI-type compounds†
Christopher J.
Adams
,
Matteo
Lusi‡
 *,
Emily M.
Mutambi
and
A.
Guy Orpen
*,
Emily M.
Mutambi
and
A.
Guy Orpen
School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
First published on 6th May 2015
Abstract
The two-step mechanochemical preparation of carbene–pyridine complexes of palladium and platinum is reported. The organometallic products, which represent a class of commercially available catalysts, are rapidly formed in excellent yield proving solvent-free synthesis to be a viable synthetic alternative even in the case of NHC-containing compounds.
Solvent-free reactions represent an opportunity to tackle difficult synthetic problems, to understand and control the behaviour of chemical species in the solid state, and to reduce the economic and environmental costs of solution-phase synthesis.1 The growing importance of the topic is highlighted in recent journal special issues.2 Most of the recent publications on the topic have as their object supramolecular (hydrogen and halogen bonded) and metalorganic complexes. Although solid state synthesis of organometallic adducts dates back almost 50 years,3 little progress has been made in this field since then.4 In order to exploit the full potential of solid state reactions, an effort is required to widen the range of chemical species produced in the solid state. This work describes the mechanochemical preparation of a commercially important class of organometallic compounds known as PEPPSIs (pyridine-enhanced precatalyst preparation stabilization and initiation).5
PEPPSI catalysts comprise a palladium(II) centre bonded to two chloride, one N-heterocyclic carbene (NHC) and one pyridine ligand. They are active in a range of organic reactions including Negishi, Buchwald–Hartwig, Kumada and Suzuki–Miyaura couplings.6–9 As well as being excellent catalysts for these reactions they possess extremely good stability, being indefinitely stable to air and surviving prolonged heating in DMSO. The synthesis of these compounds usually involves refluxing a mixture of PdCl2, K2CO3 and the hydrochloride salt of the NHC in neat 3-chloropyridine at elevated temperatures for 16 h, which gives excellent yields but requires that the excess pyridine be distilled away at the end of the procedure.5
Our group has previously reported solid-state syntheses of transition-metal coordination complexes in which target species can be formed rapidly and quantitatively by grinding together appropriate starting materials. Thus, K2MCl4 (M = Pd, Pt) will react with two equivalents of chloride salts of a range of protonated nitrogen ligands to eliminate KCl and form the tetrachlorometallate salt of the cation in a simple ion-metathesis reaction (Scheme 1).10 We have also shown that it is then possible to derive coordination complexes by solid-state dehydrochlorination of the metal-chloride salts of protonated amines and heterocyclic nitrogen bases such as pyridine, imidazole and pyrazole in the solid state with an external base such as KOH. On reaction with the base, the cation is deprotonated and coordinates to the metal, displacing a chloride ion which is eliminated as KCl (Scheme 1).10–13 This KCl is easily removed, if necessary, by extracting the metal complex product into an organic solvent. Notably, if the palladium species were to be used in catalysis (for example) then this step may not be needed: many coupling reactions eliminate KCl or KBr, so its presence as an inert contaminant added with the catalyst may be benign.
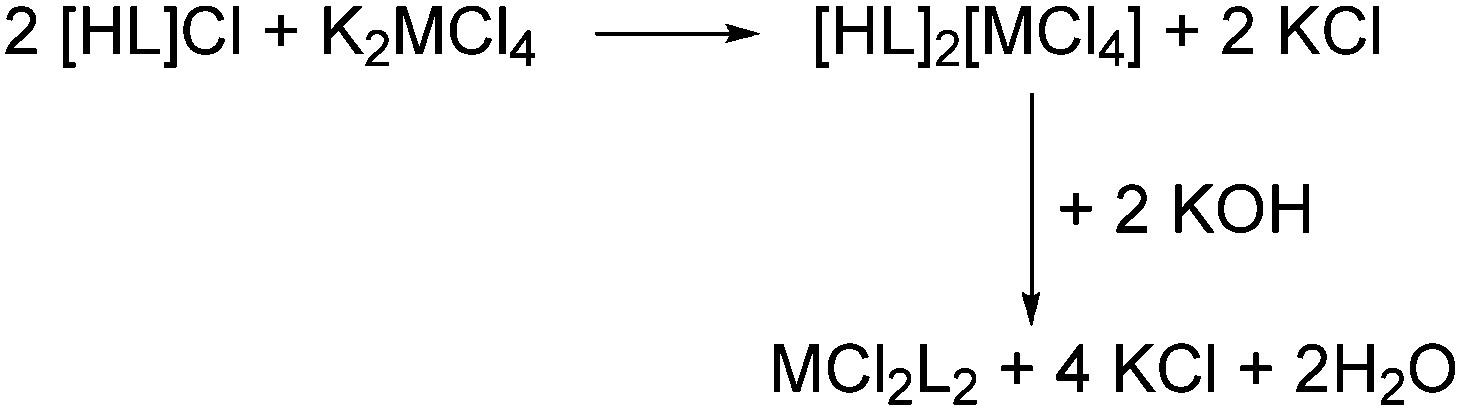 | ||
| Scheme 1 Solid-state route to coordination complexes of heterocyclic nitrogen ligands (L = pyridine, imidazole, pyrazole). | ||
Like the salts of the protonated nitrogen bases mentioned above, those of N,N′-disubstitutedimidazolium (protonated NHC) complexes form hydrogen bonds between the tetrachlorometallate anions and the cations.14 On the basis of this similarity, we conjectured that the formation of N,N′-disubstitutedimidazolium metal-chloride salts might serve to pre-arrange the anion and cation into a geometry in which the hydrogen and chlorine atoms to be eliminated on treatment with KOH are adjacent to each other. In testing this conjecture we are mindful of the difference in acidity of the hydrogen atoms in question (in, say, pyridinium [pKa ∼ 5] vs. NHC imidazolium [pKa ∼ 23]). We note, however, that the acidity of the cation may be very different at the solid state interface,15 and that a concerted reaction may be facilitated by agostic interactions between the metal and the carbon atom yielding the target organometallic adducts.16
Herein we report the solid-state synthesis of PEPPSI-type compounds. The target materials can be obtained in a two-step strategy by manually grinding together the reactants in an agate mortar.§ In the first step a three-component mechanochemical reaction involving K2MCl4, pyridinium hydrochloride (py·HCl) and one of the imidazolium hydrochloride salts – 1,3-dibenzylimidazolium chloride (IBzHCl), 1,3-bis(2,4,6-trimethylphenyl)imidazole chloride (IMesHCl), or 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPrHCl) – in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio quantitatively affords salts of general formula [HL]+[Hpy]+[MCl4]2− (M = Pd, 1; Pt, 2; L = IBz, (a); IMes, (b); IPr (c); py = pyridine) in a few minutes (Scheme 2). Powder X-ray diffraction (PXRD) patterns of the product mixtures reveals no evidence of the K2MCl4, pyridinium chloride or imidazolium chloride salts being still present, but all contain a peak at 2θ = 28.3° due to crystalline KCl, indicating the reaction has progressed (Fig. S1 and S2, ESI†).
:1:1 ratio quantitatively affords salts of general formula [HL]+[Hpy]+[MCl4]2− (M = Pd, 1; Pt, 2; L = IBz, (a); IMes, (b); IPr (c); py = pyridine) in a few minutes (Scheme 2). Powder X-ray diffraction (PXRD) patterns of the product mixtures reveals no evidence of the K2MCl4, pyridinium chloride or imidazolium chloride salts being still present, but all contain a peak at 2θ = 28.3° due to crystalline KCl, indicating the reaction has progressed (Fig. S1 and S2, ESI†).
 | ||
| Scheme 2 Synthesis of 1–6. | ||
Attempts to grow single crystals of these salts by recrystallization from common solvents were unsuccessful, giving instead (in some cases) a second series of salts: [HL] + [PdCl3py] − (3) and [HL] + [PtCl3py] − (4), which are formed by deprotonation of the pyridinium cation and the concomitant displacement of a chloride ligand at the metal. The crystal structures of [HIPr] + [PdCl3py] − (3c), [HIMes] + [PtCl3py] − (4b) and [HIPr] + [PtCl3py] − (4c) were determined from crystals grown in this way (Fig. 1). Often, two other differently coloured crystalline products were also produced in these recrystallisations; these were identified by X-ray diffraction as the bis(imidazolium) salts [Him]2[MCl4] (as red crystals) and bis(pyridine) complexes [MCl2py2] (colourless crystals) probably resulting from ligand redistribution reactions which will be reported in full elsewhere.
 | ||
| Fig. 1 Supramolecular imidazolium–chlorine H-bonds for the structures (a) 3c; (b) 4b; (c) 4c; and the organometallic complexes for (d) 5a; (e) 5b; (f) 5c. H atoms have been omitted for clarity. | ||
Deprotonation of the pyridinium fragment, as required to form 3 and 4 does not take place in the solid state, as evidenced by elemental analysis, which is not consistent with loss of HCl after the grinding and prior to crystallization (see Experimental details, ESI†). Furthermore, the powder patterns obtained following grinding do not match those calculated from the crystal structures of 3c, 4b and 4c (and nor do they contain patterns from any of the three starting materials). Therefore, it appears that the pyridinium cation is deprotonated during the dissolution/crystallization process. However the NMR spectra obtained from freshly prepared solutions of 1 and 2 (Fig. S3 to S8, ESI†) show resonances typical of the pyridinium moiety (Fig. S15, ESI†).
Notably, when the materials 1 + 2KCl and 2 + 2KCl prepared in the first step of each reaction are ground with KOH, crystalline coordination compounds trans-PdCl2(py)(NHC) (5) and trans-PtCl2(py)(NHC) (6) (NHC = N-heterocyclic carbene: IBz, (a); IMes, (b); IPr, (c)) are produced (Scheme 2). The products were characterized by spectroscopic techniques, elemental analysis and X-ray powder diffraction.
The microcrystalline products of mechanochemical reactions have microanalytical data consistent with those expected for 5 (or 6) and four equivalents of KCl. Apart from the characteristic KCl peak at 2θ = 28.3°, PXRD measurements show that the palladium and platinum organometallic complexes are isomorphous in pairs (Fig. 2). In some cases single crystals of the complexes could be grown from solution and allow comparison with the powder patterns obtained from solid-state reaction. Unsolvated crystal structures of 5a, 5b, 5c and 6a were determined (Fig. 1), as were the structures of 5c·CH2Cl2, 5c·toluene and 6b·CH2Cl2. The individual structures are unremarkable and will not be discussed in detail here; full details are available as ESI.†
 | ||
| Fig. 2 PXRD: (a) from bottom to top calculated for 5a, measured for 5a (reaction product), calculated for 6a and measured for 6a (reaction product). (b) From bottom to top calculated for 5b, measured for 5b (reaction product) and 6b (reaction product); (c) from bottom to top calculated for 5c, measured for 5c (reaction product) and 6c (reaction product). | ||
For 5a, 5b and 6a X-ray powder diffraction patterns of the products match those calculated from the crystal structures; in the case of 6b an unsolvated crystal structure is not available, but the powder pattern of the reaction product resembles that of the palladium analogue 5b.
The only case in which the PXRD pattern obtained from the as-synthesised powder does not match that calculated from the unsolvated single crystal structure is 5c suggesting that, in this case, the solid-state synthesis produces an as yet unidentified polymorph. The powder pattern for the as-synthesised 6c shows a marked similarity to that of 5c, suggesting that they are isomorphous (Fig. 2).
In conclusions this work shows that:
(1) Grinding together equimolar amounts of K2MCl4 (M = Pd, Pt), pyridinium hydrochloride and imidazolium chloride salt generates the mixed salts [HIm][Hpy][MCl4] 1 and 2, rather than the known bis(pyridinium) or bis(imidazolium) tetrachlorometallates.
(2) The pyridinium counterions in 1 and 2 spontaneously deprotonate in solution, leading to formation of the salts 3 and 4, containing imidazolium cations hydrogen-bonded to [MCl3py]− anions.
(3) Grinding 1 or 2 with two equivalents of KOH cleanly and quickly generates the PEPPSI-type mixed ligand complexes 5 and 6 in essentially quantitative yield.
Observation 1 is interesting in that three components generate a single mixed-cation product, rather than the statistical 1:2:1 distribution of bis(imidazolium):mixed salt:bis(pyridinium) compound that might be expected from a random distribution of ions. This implies that the stability of the mixed salts 1 and 2 is greater than that of the bis(imidazolium) and bis(pyridinium) salts and that the kinetics of the redistribution reactions are readily accessed by room temperature grinding. As we have found on previous occasions in related reactions, KCl is formed and it seems likely that this is a powerful driving force. It is clear that the redistribution reaction proceeds promptly in these solid state reactions without addition of solvents and thermodynamic control over the products formed is readily achieved.
Our central conjecture is that the charge-assisted hydrogen-bonding between the anions and cations is an important factor in generating the complexes, pre-organising an acidic proton near to a negatively charged chloride ion so HCl elimination is readily achieved. Although we do not have crystal structures for any of the mixed-salts 1 and 2 it seems likely (given the known predisposition of imidazolium cations to hydrogen bond to chlorometallate anions through the central CH) that the same kind of pre-organisation occurs in those species. Whether in the ground state crystal structure or readily accessed from it, this would presumably facilitate deprotonation of the pyridinium cation and place the pro-ligand and the metal fragment in proximity and so lead them to form the coordination complex when deprotonation occurs.
The complexes reported are all rapidly formed in excellent yield (practically quantitative), and can be readily separated from the KCl by-product by extraction into an organic solvent. This solvent-free synthesis is therefore a viable synthetic route to useful NHC-containing compounds. The preparations herein are amenable to scale-up and mechanization, for example by the use of ball-mills.
Notes and references
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- See CrystEngComm, Mechanochemistry and cocrystals, 2009, issue 3, pages 375 to 522; CrystEngComm Dynamic behaviour and reactivity in crystalline solids, 2011, issue 13, pages 4291 to 4438; ChemComm Mechanochemistry: fundamentals and applications in synthesis web themed issue, http://pubs.rsc.org/en/journals/articlecollectionlanding?sercode=cc&themeid=6608cfd9-e113-499d-8069-3691fd2efb77&journalname=chemical%20communications&e=1.
- L. Vaska, J. Am. Chem. Soc., 1966, 88, 5325–5327 CrossRef CAS.
- N. J. Coville and L. Cheng, J. Organomet. Chem., 1998, 571, 149–169 CrossRef CAS.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- M. G. Organ, G. A. Chass, D. C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2776–2797 CrossRef CAS PubMed.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- M. G. Organ, S. Çalimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS PubMed.
- M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. Valente, Chem. – Eur. J., 2006, 12, 4749–4755 CrossRef CAS PubMed.
- C. J. Adams, M. F. Haddow, R. J. I. Hughes, M. A. Kurawa and A. G. Orpen, Dalton Trans., 2010, 39, 3714–3724 RSC.
- C. J. Adams, H. M. Colquhoun, P. C. Crawford, M. Lusi and A. G. Orpen, Angew. Chem., Int. Ed., 2007, 46, 1124–1128 CrossRef CAS PubMed.
- C. J. Adams, M. F. Haddow, M. Lusi and A. G. Orpen, CrystEngComm, 2011, 13, 4324–4331 RSC.
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790–1795 RSC.
- P. B. Hitchcock, K. R. Seddon and T. Welton, J. Chem. Soc., Dalton Trans., 1993, 2639–2643 RSC.
- G. A. Stephenson, A. Aburub and T. A. Woods, J. Pharm. Sci., 2011, 100, 1607–1617 CrossRef CAS PubMed.
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed., 1995, 34, 2371–2374 CrossRef CAS PubMed.
- L. R. Milgrom, P. J. F. Dempsey and G. Yahioglu, Tetrahedron, 1996, 52, 9877–9890 CrossRef CAS.
- J. A. Montgomery and H. J. Thomas, J. Org. Chem., 1965, 30, 3235–3236 CrossRef CAS.
- A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS.
- M. G. Organ, C. J. O'Brien and A. B. Kantchev, US Pat., US20070073055 A1, 2007 Search PubMed.
- G. M. Sheldrick, SADABS 2.10, University of Göttingen, Germany, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic information files as well as detailed synthetic and analytical details. CCDC 1058574–1058584. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02924d |
| ‡ Current address: Chemical and Environmental Science, University of Limerick, Limerick, Ireland. E-mail: matteo.lusi@ul.ie |
| § All reagents were purchased from Aldrich and used without further purification. The ligands 1,3-dibenzylimidazolium chloride (IBz·HCl),17,18 1,3-bis(2,6-diisopropylphenyl)imidazolinium chloride (IPr·HCl),19 and 1,3-bis(2,4,6-trimethylphenyl)imidazolinium chloride (IMesHCl)20 were prepared according to the literature methods, and dried in vacuo in the oven at 60 °C (IBz·HCl) and 40 °C (IMes·HCl) and (IPr·HCl). Grinding was carried out by hand using an agate mortar and pestle. The time required for grinding varied depending on the metal, and it was noticeable that grinding of platinum salts required more time than palladium salts. NMR spectra were recorded on CDCl3 solutions and calibrated to the residual protons of the solvent (1H) or to the 13C signals of the solvent (13C). Elemental analyses were carried out by the Microanalytical Laboratory at the University of Bristol, School of Chemistry. Single crystal X-ray data were collected at 100 K on a Bruker APEX II diffractometer using Mo-Kα X-ray radiation. Data were corrected for absorption using empirical methods (SADABS)21 based upon symmetry-equivalent reflections combined with measurements at different azimuthal angles. Crystal structures were solved and refined against all F2 values using the SHELXTL suite of programs.22 Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters. The Crystallographic Information Files are available from the CCDC 1058574–1058584. All crystalline phases were analyzed at room temperature by powder X-ray diffraction on a Bruker D8 diffractometer using Cu-Kα X-radiation. |
| This journal is © The Royal Society of Chemistry 2015 |