
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Red, far-red, and near infrared photoswitches based on azonium ions†
M.
Dong
a,
A.
Babalhavaeji
a,
M. J.
Hansen
a,
L.
Kálmán
b and
G. A.
Woolley
 *a
*a
aDepartment of Chemistry, University of Toronto, Toronto, M5S 3H6, Canada. E-mail: awoolley@chem.utoronto.ca
bDepartment of Physics, Concordia University, Montreal, Quebec H4B 1R6, Canada
First published on 8th July 2015
Abstract
Azonium ions formed by p-amino substituted azo compounds with both ortho- and meta-methoxy substituents exhibit strong absorbance in far-red and near infrared spectral region. The compounds undergo robust photoswitching in aqueous solution and exhibit a range of thermal relaxation rates from 10 μs–100 ms.
Molecules that undergo reversible light driven changes in structure are of increasing interest for applications in fields ranging from molecular electronics to neurobiology.1–3 For biological applications in particular, considerable efforts have been made to produce molecules that absorb in the red region of the visible spectrum since these wavelengths enable much greater tissue penetration and less phototoxicity than shorter wavelengths.4–9 Ideally, however, molecular photoswitches designed for in vivo use should operate in the near infrared (IR) optical window.6,10 The edges of the near IR window depend on the tissue in question, but are generally set by haemoglobin absorption at shorter wavelengths and water absorption at longer wavelengths. The best penetration is often observed near 730 nm.11 Although some near IR absorbing azo compounds are known, these are complex, flexible, molecules that would be difficult to apply as photoswitches.12,13 Very recently Aprahamian and colleagues described near IR absorbing photoswitches based on BF2-modified azo compounds.10 These compounds showed excellent photochemical characteristics, however, their tendency to hydrolyse in aqueous solution presents a serious drawback to their application in a biological context.
We recently described the azonium ion 1 that absorbed red light and underwent trans–cis photoisomerization.14 It relaxed to the trans isomer in the dark on the timescale of seconds so that pulses of red light could be used to drive multiple isomerization cycles. Resonance stabilization of the azonium cation together with intramolecular H-bonding between the azonium proton and methoxy groups ortho to the azo unit meant that the trans azonium species was present at pH 7 in aqueous solution, whereas typically azonium ions are only formed from aminoazobenzenes at pH < 3.5.15–17 The geometry of the cis isomer prevents formation of this intramolecular H-bond so that the cis azonium pKa is lower than that of the trans isomer.14 As a result, depending on the pH, trans to cis isomerization is accompanied by proton dissociation to produce the neutral cis species, thereby slowing thermal reversion to the trans form. This feature is useful for the photochemical production of a large fraction of this cis isomer to control molecular targets. Since compound 1 acts as an effective red light switch that can operate in biological milieu, we used it as a starting point to try to design azonium ion based switches that would operate in the near IR.
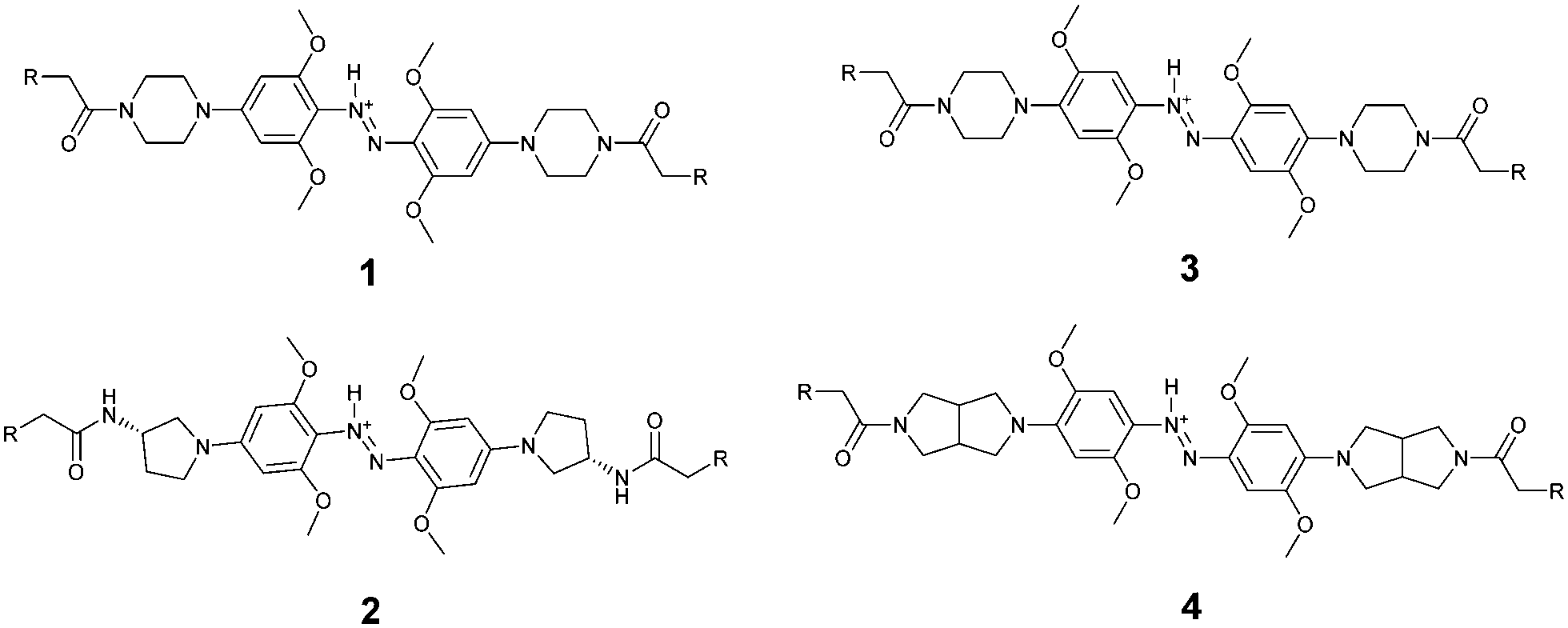
There is a rich literature describing substituent effects on the absorption wavelengths of azobenzenes and, to a lesser extent, the corresponding azonium ions.18 We wished to keep at least one methoxy substituent in an ortho position to preserve the possibility of intramolecular H-bonding of the trans azonium ion. We therefore considered changes at para and meta positions. Varying the nature of the amine in the para position alters the degree to which the p-amino nitrogen lone pairs delocalize into the ring system.19 A second phenomenon, called the distribution rule of auxochromes was recognized early by Kaufmann20 and described in detail by Wizinger.21 This rule predicts that a second auxochrome (e.g. a methoxy substituent) in a meta position should shift absorbance to longer wavelengths. The phenomenon is not explained using heuristic rules of resonance and historically was the subject of much discussion,21,22 but nowadays is predicted by standard quantum chemical calculations. In an effort to produce photoswitches operating at longer wavelengths than (1), we decided to examine the role of the methoxy substitution pattern as well as the nature of the para amino group in the photochemical behaviour of these azonium-based photoswitches.
Compounds 2, 3, and 4 were designed to preserve the possibility of H-bonding of the azonium proton to an ortho-methoxy group in the trans isomer (as in 1) but to have longer wavelength absorption due to the presence of better para donating substituents (2vs.1, 4vs.3) or an ortho–meta methoxy substitution pattern (3 and 4vs.1 and 2). In addition to affecting the wavelength of absorption, these changes are expected to alter the pKa of the azonium ion formed in each case. Calculations were performed using density functional methods (B3LYP (6-31++G**)) to optimize geometry and TDDFT to calculate absorption wavelength maxima (see ESI†). These computational methods have been used successfully with related compounds.23,24 TDDFT calculations predict that these compounds should absorb at longer wavelengths than 1 (Table 1). Calculating effects of the substitution patterns on pKas is problematic25 and was not attempted here.
| Compound | Predicted λmax (trans neutral form) (nm) | Observed λmax (trans neutral form) (nm) | Predicted λmax (trans azonium form) (nm) | Observed λmax (trans azonium form) (nm) | ε (λmax) (trans azonium form) (M−1 cm−1) | Apparent pKa of azonium ion |
|---|---|---|---|---|---|---|
| 1 | 522, 384 | 460, 365 | 545 | 563 | 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
7.5 |
| 2 | 518, 384 | 491, 410 | 544 | 610 | 51000 |
8.9 |
| 3 | 497 | 490 | 631 | 630 | 40000 |
2.6 |
| 4 | 485 | 517 | 634 | 670 | 37000 |
5.4 |
Compound 2 was synthesized in a manner analogous to that reported previously for 1 except that an amino-pyrrolidine unit replaced the piperazine unit in 1 (see ESI† for details). Compounds 3 and 4 were synthesized as outlined in Scheme 1. Commercially available 1-bromo-2,5-dimethoxybenzene was nitrated to give compound 5. Reduction using hydrazine hydrate and Pd/C, produced compound 6. The key intermediate 7 was generated by treating 6 with CuBr and pyridine in toluene.
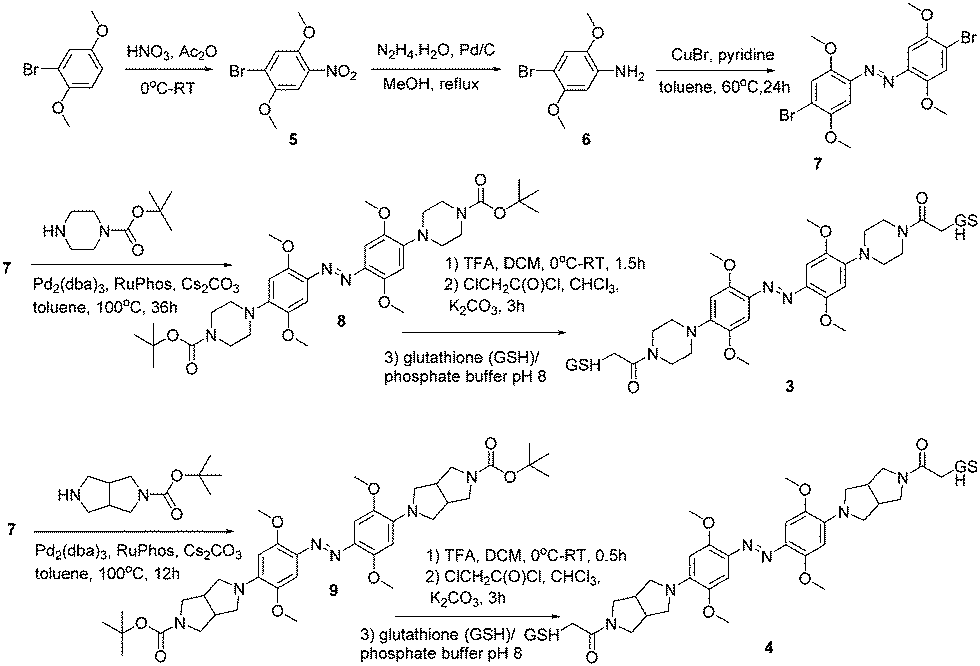 | ||
| Scheme 1 | ||
To produce 4 we used a bicyclic pyrrolidine to preserve the symmetry of the compound and to maximize the end-to-end distance change upon trans to cis isomerization; both these features are important for applying these compounds as cross-linkers for peptides and proteins.26 The bicyclic pyrrolidine version of 2 proved to be insoluble so that it was replaced by the monocyclic amino-pyrrolidine in that case. These different pyrrolidine moieties are expected to behave similarly in terms of electron donation to the azo system. In each case, the compound was linked at both ends to the short tripeptide glutathione (GSH). This confers water solubility on the photoswitch and also provides a test of the operational sensitivity of the compound to reduction by thiols. While azonium ions can, in general, be reduced by thiols14,24 the rate of reduction depends on the pH and thiol concentration which vary widely depending on the particular biological environment in question.27 In practice, we find that if a GSH adduct can be readily synthesized, the rate of reduction is slow enough that the compound can be used under typical physiological conditions.
The absorption properties of compounds 2, 3, and 4 were examined in aqueous buffer as a function of pH. These compounds were found to be stable for weeks at room temperature in aqueous solutions near neutral pH, an important requirement for biological applications. Fig. 1 shows UV-Vis absorption spectra for each newly synthesized compound together with those obtained previously for compound 1. In the high pH limit, the spectrum in each case is assumed to correspond to that of the neutral (unprotonated) species. As the pH is lowered, a species with long wavelength absorption appears and this is assumed to be the singly protonated azonium ion. Contributions to the spectra from doubly protonated species (azonium plus ammonium) are indicated by dashed grey lines in Fig. 1 where these dominate the spectra. Apparent values for the pKas of the azonium ions are indicated in each case, obtained by fitting the observed spectra as function of pH (see the ESI†). The molar extinction coefficients of the trans azonium forms of 1–4 are collected in Table 1 (see ESI† for methods). These are similar to that found for methyl orange (55000 M−1 cm−1).28 TDDFT calculations also predict large oscillator strengths for these compounds (see ESI†).
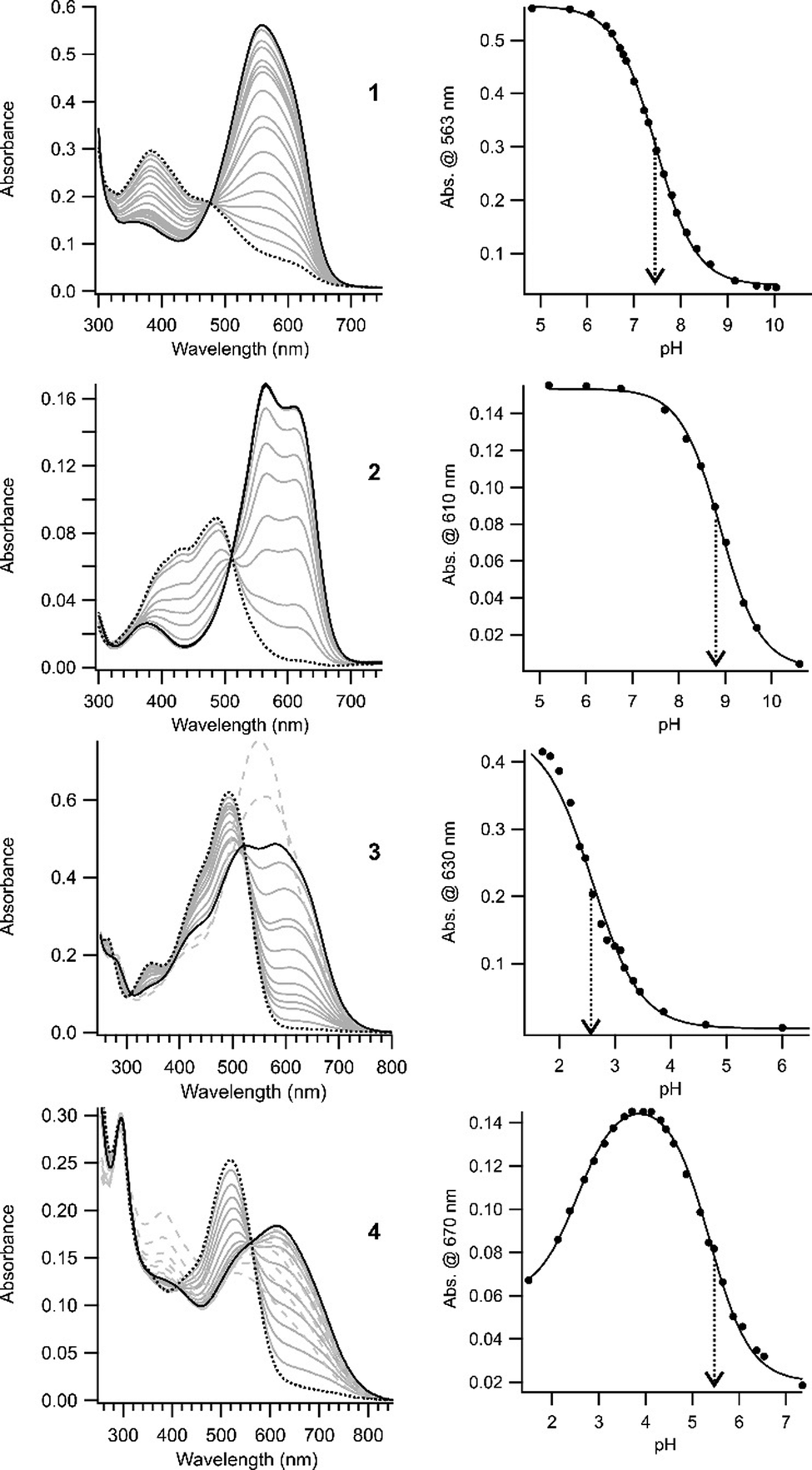 | ||
| Fig. 1 (left) UV-Vis spectra of the trans forms of compounds 1–4 (linked to GSH) in aqueous buffer as a function of pH. In each case, the spectrum at the high pH limit is shown as a black dotted line. Lowering the pH produces the azonium ion (shown as a series of grey solid lines) until a maximum amount of the azonium ion is obtained (solid black line). Further decreases to the pH produce doubly protonated species (azonium plus ammonium). The spectra of these species are indicated by dashed grey lines. (right) pH dependencies of the azonium ions formed in each compound. Solid lines represent the fits to eqn (1) for compounds 1, 2, and 3, and eqn (2) for compound 4. The dotted arrows mark the pKas of the azonium ions (see ESI†). | ||
Increasing the donating ability of the para amino substituent moves absorption from the red region into the far-red (compound 2vs.1). Introducing the ortho–meta substitution pattern produces azonium ions absorbing in the far-red (3) and near IR (4). Compound 4 shows significant absorbance in the near IR window at 730 nm.
Whether the azonium ion is present at physiological pHs near ∼7, depends on the pKa. The apparent pKas of these compounds are collected in Table 1. Increasing the donating ability of the para amino substituent increased the pKa of the trans azonium ion of 2 to 8.9 (compared to 7.5 for 1). A consequence of this change is that the azonium ion dominates at physiological pHs near ∼7 so that red and far-red wavelengths are absorbed strongly. Replacing two ortho methoxy groups with an ortho–meta substitution pattern (e.g.3vs.1 or 4vs.2) causes a significant drop in the pKa of the corresponding trans azonium ions, particularly in the case of 3. The removal of an ortho methoxy group removes a possible resonance contributor for stabilization of the positive charge. In addition, it removes a potential H-bond acceptor; however, calculations suggest that H-bonded species still predominate (see ESI†) suggesting the latter is not a major reason for the drop in the azonium pKa. Instead, the pKa drop may result from the presence of meta methoxy group which forces the para piperazino substituent of 3 to rotate so that the para piperazino nitrogen atom is significantly less able to act as an electron donor. Calculated structures of 3 (Fig. S1, ESI†) confirm that the piperazino units are highly twisted. Consistent with this proposal, the less sterically demanding pyrrolidino substituent (compound 4) shows a significantly higher pKa (Table 1).
We then examined the photoswitching behaviour of this series of compounds using laser flash photolysis techniques. Hundreds of rounds of trans–cis photoisomerization occurred in each case and photocyclization of the type observed with simple azonium ions by Lewis29 and discussed by Mallory30 does not seem to occur with these compounds. The rate of cis to trans thermal relaxation increases as the pH is lowered because cis azonium species relax significantly more quickly than their neutral counterparts.31Fig. 2 shows the observed first order rate constants for thermal cis-to-trans relaxation as a function of pH for compounds 1–4 in aqueous solution. In general, the rate constants are bounded at the low pH end by the intrinsic relaxation rate of the cis azonium ion and at the high pH end by the intrinsic relaxation rate of the neutral species in each case. Not all pH/rate combinations were experimentally accessible. At a given pH the observed rate reflects both the intrinsic thermal relaxation rate as well as the pKa of the cis azonium ion.14 At physiological pH (∼7) the tetra-ortho methoxy substituted species 1 and 2 show cis lifetimes on the order of 1 s and ∼100 ms respectively.
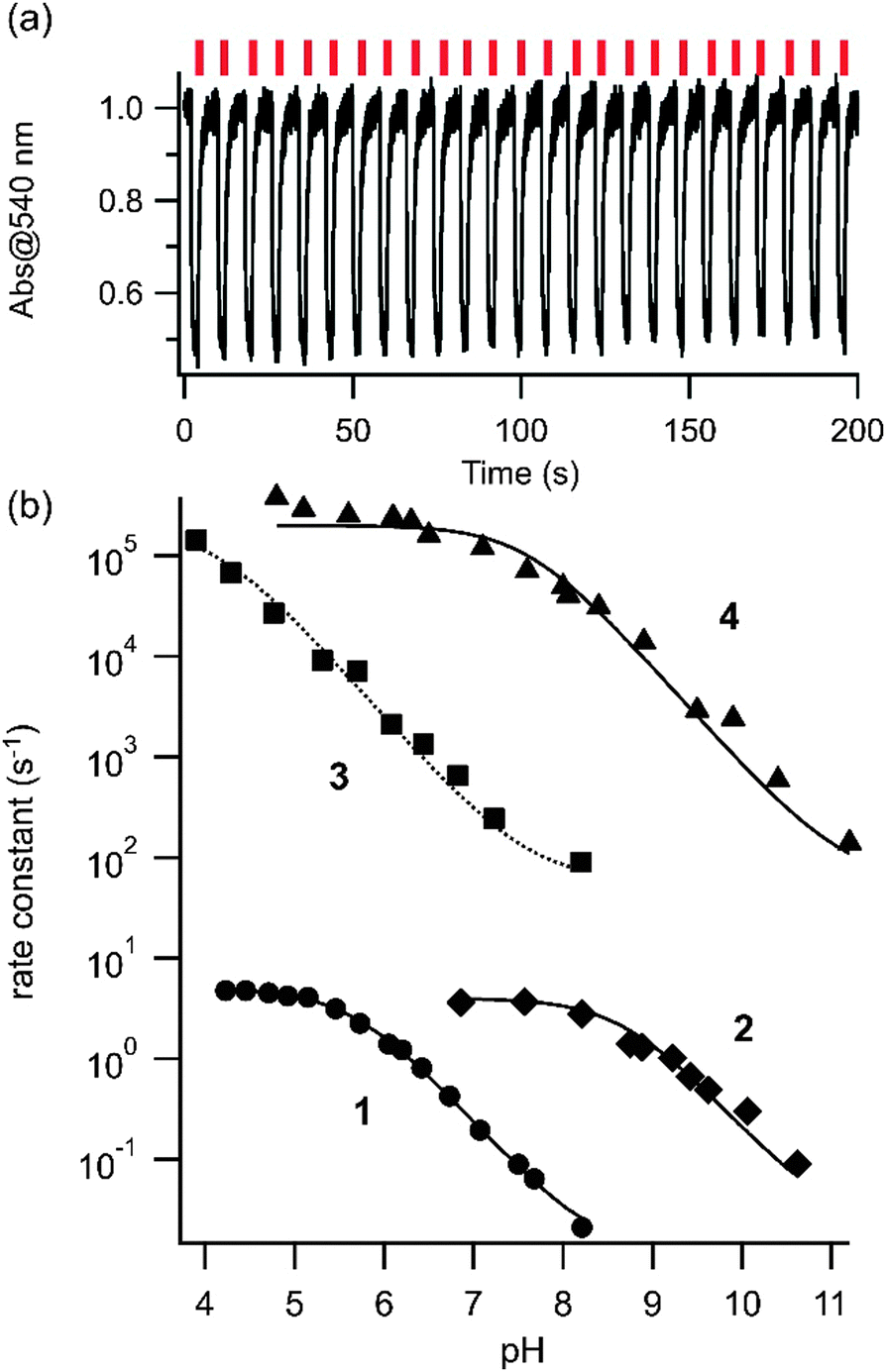 | ||
| Fig. 2 (a) Far-red photoisomerization of 2. Absorbance changes observed with pulses of 660 nm light (aqueous buffer, pH 8.8, 20 °C). (b) Observed rate constants for thermal cis-to-trans relaxation of compounds 1–4 as a function of pH in aqueous buffer. The kinetic traces of thermal relaxation were fit to single exponential decay functions to obtain rate constants (see ESI†). Circles, diamonds, squares, and triangles show the observed rate constants for compounds, 1, 2, 3, and 4, respectively. The lines represent the fit to eqn (3) yielding pKa values of the cis azonium ions of 5.7, 8.6, 4.1, and 7.6 respectively (see ESI† for details). | ||
Compound 2 thus functions as a far-red photoswitch that operates at physiological pHs. Compounds 3 and 4 have lifetimes of ∼1 ms and 10 μs respectively at pH 7, however only compound 4 has significant near IR absorbance at this pH. Compound 4 could thus function as a near IR switch with rapid (10 μs) thermal relaxation. While rapid thermal relaxation is ideal for certain applications32,33 slower relaxation permits substantial fractions of the cis isomer to be produced at low irradiation powers and is useful when a large degree of photo-control of single target biomolecules is desired.1 The thermal relaxation data make it clear that substitution at all four ortho positions is necessary to substantially slow the thermal back reaction. Thus, if a compound with near IR absorbance together with a slow thermal relaxation rate is desired, our data predict that substitution of all four ortho positions in addition to a meta position will be required.
Substituted azonium ions can function as water-stable photoswitches that operate with red, far-red and near IR light. Whereas tetra-ortho methoxy substitution is required for slow thermal reversion, a meta methoxy substitution pattern enhances the long wavelength absorbance of azonium ions. A meta methoxy substitution places steric constraints on the nature of the para substituent so that a pyrrolidino group is best able to act as an electron donor in this position. Varying these substituents allow the construction of azonium-based photoswitches with different degrees of far-red absorbance, thermal relaxation rate and pKa tuned for desired applications.
We are grateful to Mightex Inc. for the loan of red, far-red and near IR emitting high power LED sources. This work was supported by the Natural Sciences and Engineering Research Council of Canada.
Notes and references
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- Molecular switches, ed. B. Feringa and W. R. Browne, Wiley-VCH, 2011 Search PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascon-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, E. Ferrer, R. Alibes, F. Busque, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS PubMed.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686 CrossRef CAS PubMed.
- J. Broichhagen, J. A. Frank, N. R. Johnston, R. K. Mitchell, K. Smid, P. Marchetti, M. Bugliani, G. A. Rutter, D. Trauner and D. J. Hodson, Chem. Commun., 2015, 51, 6018–6021 RSC.
- H. Nishioka, X. G. Liang, T. Kato and H. Asanuma, Angew. Chem., Int. Ed. Engl., 2012, 51, 1165–1168 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190–13193 CrossRef CAS PubMed.
- L. V. Wang and H.-I. Wu, Biomedical optics: Principles and imaging, Wiley, 2007 Search PubMed.
- Y. Li, B. O. Patrick and D. Dolphin, J. Org. Chem., 2009, 74, 5237–5243 CrossRef CAS PubMed.
- K. A. Bello and J. Griffiths, Chem. Commun., 1986, 1639–1640 RSC.
- S. Samanta, A. Babalhavaeji, M. X. Dong and G. A. Woolley, Angew. Chem., Int. Ed. Engl., 2013, 52, 14127–14130 CrossRef CAS PubMed.
- S. K. Guha and O. P. Jha, J. Indian Chem. Soc., 1968, 45, 365 CAS.
- S. Stoyanov, L. Antonov, T. Stoyanova and V. Petrova, Dyes Pigm., 1996, 32, 171–185 CrossRef CAS.
- T. Stoyanova, S. Stoyanov, L. Antonov and V. Petrova, Dyes Pigm., 1996, 31, 1–12 CrossRef CAS.
- E. Sawicki, J. Org. Chem., 1957, 22, 1084–1088 CrossRef CAS.
- G. Hallas, R. Marsden, J. D. Hepworth and D. Mason, J. Chem. Soc., Perkin Trans. 2, 1984, 149–153 RSC.
- H. Kauffmann and W. Kugel, Ber. Dtsch. Chem. Ges., 1911, 44, 2386–2389 CrossRef CAS PubMed.
- K. Kokkinos and R. Wizinger, Helv. Chim. Acta, 1971, 54, 330–334 CrossRef CAS PubMed.
- R. Wizinger, Chimia, 1965, 19, 339–350 CAS.
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef CAS PubMed.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Chem. Commun., 2013, 49, 10314–10316 RSC.
- K. S. Alongi and G. C. Shields, Annu. Rep. Comput. Chem., 2010, 6, 113–118 CAS.
- A. A. Beharry, T. Chen, M. S. Al-Abdul-Wahid, S. Samanta, K. Davidov, O. Sadovski, A. M. Ali, S. B. Chen, R. S. Prosser, H. S. Chan and G. A. Woolley, Biochemistry, 2012, 51, 6421–6431 CrossRef CAS PubMed.
- X. Jiang, Y. Yu, J. Chen, M. Zhao, H. Chen, X. Song, A. J. Matzuk, S. L. Carroll, X. Tan, A. Sizovs, N. Cheng, M. C. Wang and J. Wang, ACS Chem. Biol., 2015, 10, 864–874 CrossRef CAS PubMed.
- J. F. Boily and T. M. Seward, J. Solution Chem., 2005, 34, 1387–1406 CrossRef CAS.
- G. E. Lewis, J. Org. Chem., 1960, 25, 2193–2195 CrossRef CAS.
- F. B. Mallory and C. W. Mallory, Org. React., 2005, 30, 1–456 Search PubMed.
- A. M. Sanchez, M. Barra and R. H. de Rossi, J. Org. Chem., 1999, 64, 1604–1609 CrossRef CAS PubMed.
- J. Garcia-Amoros, M. Diaz-Lobo, S. Nonell and D. Velasco, Angew. Chem., Int. Ed. Engl., 2012, 51, 12820–12823 CrossRef CAS PubMed.
- J. Vapaavuori, A. Goulet-Hanssens, I. T. S. Heikkinen, C. J. Barrett and A. Priimagi, Chem. Mater., 2014, 26, 5089–5096 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, quantum chemical calculations, and photoisomerization measurements. See DOI: 10.1039/c5cc02804c |
| This journal is © The Royal Society of Chemistry 2015 |