
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly nanoporous silicas with pore apertures near the boundary between micro- and mesopores through an orthogonal self-assembly approach†
Koki
Muraoka
a,
Watcharop
Chaikittisilp
*a,
Yutaka
Yanaba
b,
Takeshi
Yoshikawa
b and
Tatsuya
Okubo
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: watcha@chemsys.t.u-tokyo.ac.jp; okubo@chemsys.t.u-tokyo.ac.jp
bInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo, 153-8505, Japan
First published on 21st May 2015
Abstract
Nanoporous silicas having some periodicity, high surface area (up to 1230 m2 g−1), and pore diameters near the boundary between micro- and mesopores are synthesized using aromatic compounds bearing anionic end-groups as novel structure-directing agents (SDAs) that can facilitate multiple interactions between SDAs, co-SDAs and silica frameworks orthogonally.
Silica-based nanoporous materials, including zeolite and mesoporous silica, are one of the most important class of nanoporous materials mainly because they have been practically used in diverse fields as adsorbents, catalysts, catalyst supports, low-n and low-k materials, drug delivery carriers, and so on.1 By general definition, zeolite has its pore diameters in the microporous scale (less than 2 nm), while the pore sizes of mesoporous silica are 2–50 nm. However, silica-based nanoporous materials with pore sizes near the boundary of micro- and mesoporous scales are still rare.2 This micro–meso transition length scale is of great scientific significance because adsorption behavior can change from monolayer to multilayer modes, probably making the classical adsorption theories become inapplicable.3 From a technological viewpoint, moreover, one can expect that different physico-chemical properties can arise from this transition region, thereby providing opportunities for new applications.
Two main approaches can be conceived to achieve silica-based materials having porosity at the micro–meso transition. One is enlarging the micropores of zeolite, while the other is shrinking the pore size of mesoporous silica. The former is exceptionally difficult as such extra-large-pore zeolites tend to be thermodynamically less stable than their counterparts with smaller pores.4 The pore sizes of zeolites are generally limited to 1.3 nm,5 although there are a few exceptions.2a,b By the latter approach, a straightforward strategy is to shorten the length of the alkyl or aliphatic chains of structure-directing agents (SDAs), also called surfactants, used to synthesize mesoporous silica, which roughly determines the size of mesopores.6 It is, however, still challenging to achieve the silica materials with uniform pore sizes at and below 2 nm because the shortened alkyl chains can somewhat lose the self-assembling ability.
Orthogonality, originally termed in mathematics, is defined as the relation of two or more non-overlapping, independent objects or functions. In chemistry, materials science and life science, an orthogonal self-assembly represents hierarchical self-assembling processes that involve multiple interactions, being independent but working together cooperatively in a synergistic way. The orthogonal approach provides a powerful tool to create more complex matters by utilizing the combination of different interactions, being covalent or non-covalent, where modifications can be made to a particular motif without affecting the others.7 This approach has been widely employed to achieve hierarchical supramolecular assemblies and soft materials. It is also noteworthy that some other supramolecular approaches such as micelle assembly have been successfully applied to synthesize nanoporous architectures with unique properties.8 We have deemed that the orthogonal assembly may overcome the problems in reducing the pore sizes of mesoporous silica to around 2 nm as the orthogonal interactions of SDA–SDA, SDA–silica, and silica–silica can improve the assembly even for the SDAs with the shortened alkyl chains.
Herein, we report the synthesis of nanoporous silicas having pore apertures near the micro–meso boundary by using novel SDAs that can facilitate multiple, orthogonal interactions (see Fig. 1). To enhance the SDA–SDA interaction, disk-like molecules with rigid, π-conjugated cores have been considered as SDAs because these molecules can interact with each other via π–π interactions and thus form one-dimensional stacks. Although ordered columnar assemblies can be formed from molecular disks with, for example, phthalocyanine, triphenylene, and N-donor ligand–metal complex cores,9 their molecular sizes are too large for the formation of nanopores near the micro–meso boundary. Therefore, a benzene core, the smallest moiety that can form columnar assemblies through π–π stacking,10 was selected.
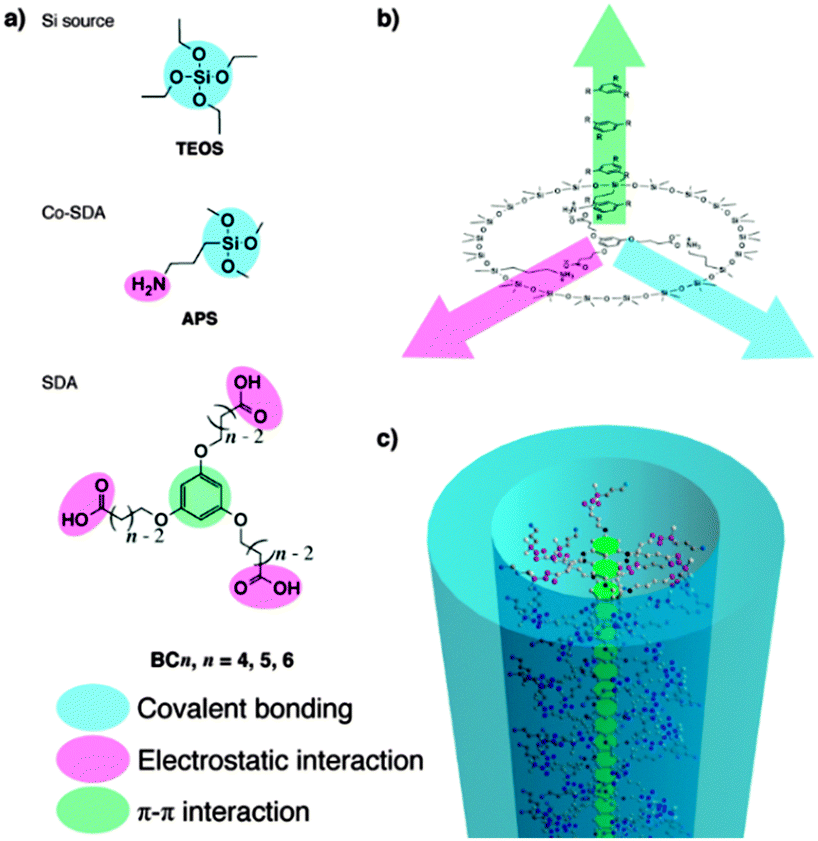 | ||
| Fig. 1 (a) Molecules and (b) schematic representation of the orthogonal interactions used in this work. (c) Nanoporous silica built through an orthogonal self-assembly approach. | ||
The interaction between SDA and silica depends on side chains attaching to the core, namely hydrogen-bonding interaction (for nonionic alkyl polyoxyethylene ether side chains)9 or electrostatic interaction (for cationic10 and anionic side chains). Because of its strength, electrostatic interaction is more preferable. Cationic SDAs with a benzene core and long alkyl side chains have been used to synthesize mesoporous silicas.10 When the cationic alkyl side chains are shortened, however, synthesis of nanoporous silica with smaller, uniform pore sizes is unsuccessful likely because of the weak interactions between positively charged, basic ammonium side chains and negatively charged, weakly acidic silanol (SiOH) groups.10 Alternatively, we employed here the concept of anionic SDA–co-SDA interaction, established by Yokoi, Che and Tatsumi for the synthesis of anionic surfactant-templated mesoporous silicas (AMSs),11 in which we have hypothesized that this can critically improve the formation of nanoporous silica because the acidity of anionic SDA (pKa ∼ 3–6) is stronger than that of silanol (pKa ∼ 7).11e,12 Based on the aforementioned consideration, novel anionic SDAs (BCn, Fig. 1a) were designed and synthesized (see experimental details and characterization in the ESI,† Fig. S1), while TEOS (tetraethyl orthosilicate) and APS (3-aminopropyltrimethoxysilane) were used as silica source and co-SDA, respectively. The molecular sizes of BCn together with APS can create the pore sizes near the micro–meso boundary, being 1.5–2.5 nm, as computed by DFT optimization (B3LYP/6-31G(d) level) (see Fig. S2, ESI†). As a co-SDA, APS can interact with SDAs via electrostatic interaction and can covalently co-condense with TEOS to form silica frameworks.
Learning from natural and biological systems that can use several weak interactions (e.g., π–π stacking, hydrogen-bonding, and electrostatic charge-matching) efficiently in aqueous media at neutral pH and moderate temperature,13 nanoporous silicas were synthesized at 60 °C from the starting mixtures with the molar composition of 0.05 BCn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :xAPS:(1 − x) TEOS:90 H2O, where x = 0–0.30. The as-made white powder was either calcined at 550 °C for 6 hours or extracted with ammonium acetate in ethanol11d (see details in the ESI†). The pH of the starting mixture depends on the amount of APS (i.e., an x value), but being close to neutral.
:xAPS:(1 − x) TEOS:90 H2O, where x = 0–0.30. The as-made white powder was either calcined at 550 °C for 6 hours or extracted with ammonium acetate in ethanol11d (see details in the ESI†). The pH of the starting mixture depends on the amount of APS (i.e., an x value), but being close to neutral.
The co-condensation of APS and TEOS was confirmed by solid-state 29Si magic-angle spinning (MAS) NMR spectroscopy (Fig. S3, ESI†). The signals arising from Q4, Q3, Q2, and T3 are observed at δ = −111, −101, −91, and −66, respectively (Qa: Si(OSi)a(OH)4−a; Tb: CSi(OSi)b(OH)3−b), suggesting that both APS and TEOS co-condense to form silica products. For the sample prepared with BC6 (x = 0.18), the peak ratio of (Q2 + Q3 + Q4)/T3, being 4.1, was slightly altered from the initial molar ratio (TEOS/APS = 4.6) probably because the ethoxy group of TEOS is less reactive than the APS methoxy group.
Solid-state 1H–13C cross-polarization (CP)/MAS NMR and UV-Vis spectroscopies were used to investigate the presence of organic groups (APS and BCn) in the as-made products. The as-made silica-BC6 sample showed the NMR peaks corresponding to the 3-aminopropyl moiety at δ = ca. 10, 22, and 42, in addition to the peaks from BC6 (Fig. S4, ESI†), confirming that both APS and BCn are incorporated in the as-made sample. The molar ratios of BCn/APS in the as-made sample were almost identical to those of the starting mixtures, as determined by elemental analyses. In UV-Vis spectra (Fig. S5, ESI†), the peaks around 240 nm can be assigned to the π–π stacking of benzene rings of BCn.10,14 Compared to the BCn powder, these peaks are blue-shifted when BCn was incorporated into the as-made samples, suggesting that the arrangements of BCn in the as-made samples are predominant by face-to-face H-aggregation. In the powder form of molecules, BCn can arrange in both face-to-face and end-to-end (J-aggregation) modes due to π–π interaction of benzene cores and hydrogen-bonding interaction of carboxylic end-groups, respectively. In the as-made samples, in contrast, BCn is likely piled up to form one-dimensional arrangement through π–π interaction, causing the blue-shift in UV-Vis spectra.
Powder X-ray diffraction (XRD) patterns of the products synthesized using SDAs having different lengths of alkyl side chains at varied amounts of APS are shown in Fig. 2. The silica products obtained at x = 0.18, in which the molar ratio of COOH/NH2 is close to 1, show broad XRD peaks with a d-spacing of ca. 3 nm, suggesting that they likely possess some periodicity.15 Among BCn, the XRD peaks are more pronounced when the products were synthesized using BC6. The products synthesized without BCn or APS do not show any XRD peaks (see Fig. S6 and S7 in the ESI,† respectively). These results suggest that the presence of both BCn and APS in the synthesis mixture at the optimal ratio (COOH/NH2 ∼ 1) is crucial to promote the electrostatic interaction of SDAs and co-SDA, thereby enhancing the periodicity of the products.
 | ||
| Fig. 2 Powder XRD patterns of the as-made (black line) and calcined (grey line) products synthesized with SDAs having different lengths of alkyl side chains at (a) x = 0.10, (b) x = 0.18, and (c) x = 0.30. | ||
The SEM image (Fig. S8, ESI†) shows that the product has an irregular shape with non-uniform particle size. A typical high-resolution (HR) TEM image of the calcined silica-BC6 sample (x = 0.18) shown in Fig. 3 is translucent, which is indicative of porous characteristics. The pore sizes were estimated to be about 2 nm. As shown in Fig. 3 (inset), the fast Fourier transform (FFT) of the selected area shows a ring pattern, indicating that the selected area possesses some periodicity. Additional HRTEM images are shown in Fig. S9, ESI.†
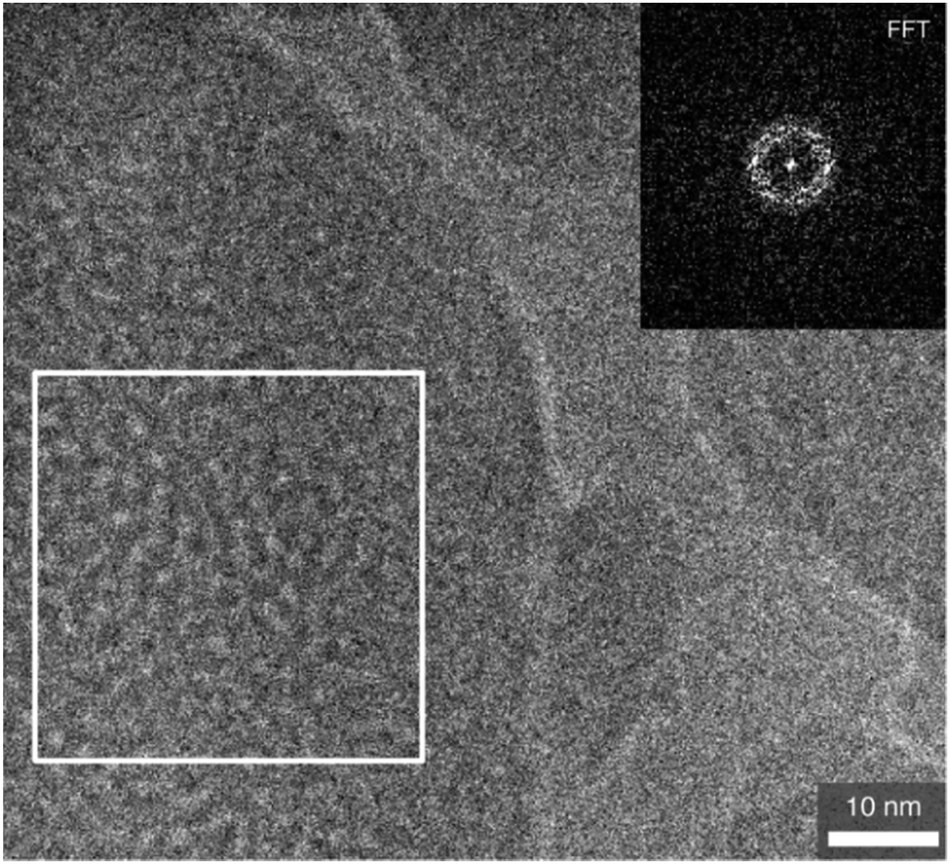 | ||
| Fig. 3 High-resolution TEM image of the calcined silica-BC6 sample. The inset shows the FFT of the marked area (white frame). | ||
Nitrogen adsorption–desorption isotherms and their corresponding pore size distributions calculated by a non-local density functional theory (NL-DFT) of the calcined samples are depicted in Fig. 4 (x = 0.18) and Fig. S10 (x = 0.10 and 0.30; ESI†), together with apparent specific surface areas (calculated by the Brunauer–Emmett–Teller (BET) method), micropore volumes (calculated by the NL-DFT method), and total pore volumes (at P/P0 = 0.99). At x = 0.18, the sample prepared with BC4 shows sharp uptakes at low relative pressure (P/P0 < 0.05) with slightly increasing uptakes at higher relative pressure, indicating that it is a microporous material. In contrast, the samples synthesized with BC5 and BC6 show considerably sharp uptakes at P/P0 up to 0.15, indicating that their nanopores lie on the boundary between micro- and mesoporous regions. Pore diameters were evaluated by the NL-DFT method, being consistent with the molecular sizes of APS and BCn. The pore size distribution of the sample derived from BC4 is centered at 1.7 nm, while the samples from BC5 and BC6 have pore diameters centered at 2.2 nm. Notably, the pore size distributions become slightly broader with extended carbon chains connected to the benzene core, probably due to the increasing flexibility of the side chains. Similar trends were also observed when x = 0.10 and 0.30, with the pore diameters of ca. 1.5–3 nm, depending on synthesis conditions and SDAs (Fig. S10, ESI†).
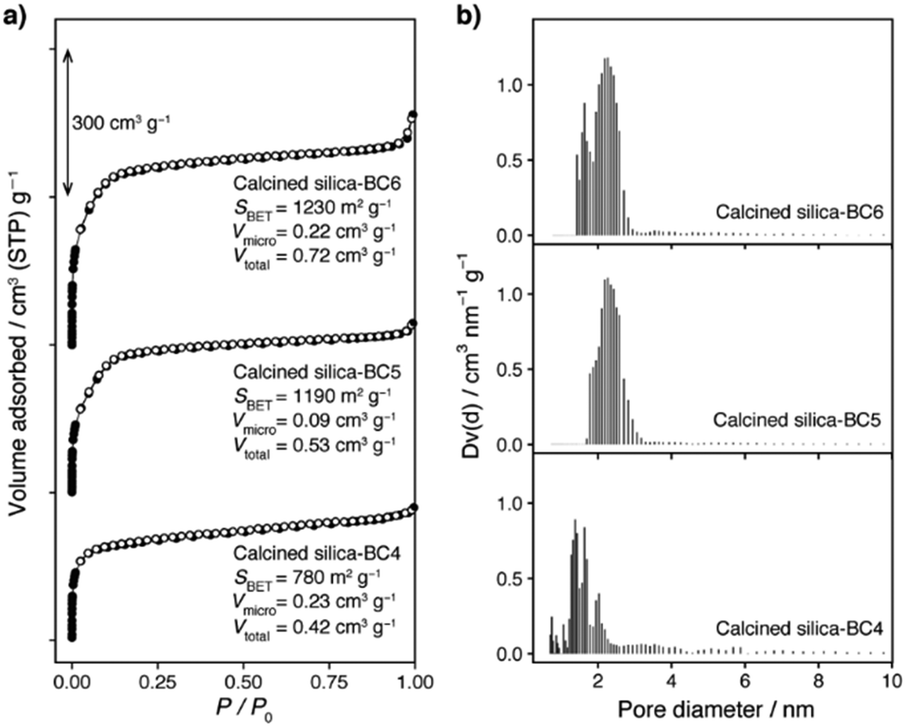 | ||
| Fig. 4 (a) Nitrogen sorption isotherms (filled and empty symbols representing adsorption and desorption branches, respectively) and (b) the corresponding histograms of NL-DFT pore size distributions of the calcined products synthesized at x = 0.18 with SDAs having different lengths of alkyl side chains. In (a), the isotherms of calcined silica-BC5 and calcined silica-BC6 were offset by 300 and 600 cm3 g−1 STP, respectively. | ||
It is now accepted that argon adsorption can give more accurate estimation of the pore size distribution than that calculated from nitrogen adsorption.16 To confirm the pore size distributions of the resulting materials, argon adsorption was also measured, giving the pore size distributions akin to those obtained from nitrogen adsorption with slightly larger diameters (ca. 0.1–0.2 nm) (Fig. S11, ESI†). It should be noted that the estimation of pore size distribution is somewhat sensitive to the pore structure,16 and therefore further characterization of pore structures (e.g., 1-dimensional cylindrical or 3-dimensional interconnected pores) should be conducted in the future.
The apparent specific BET surface area, micropore volume, and total pore volume are increased as the length of side carbon chains increased (see Fig. 4 and Fig. S10, ESI†). The highest values are observed for the silica synthesized at x = 0.18 with BC6, being 1230 m2 g−1 (surface area), 0.22 cm3 g−1 (micropore volume), and 0.72 cm3 g−1 (total pore volume). To the best of our knowledge, these results are the highest values reported for AMSs.11 It is noteworthy that the products obtained without the addition of APS are mesoporous silica with lower BET surface areas and very broader pore size distributions (Fig. S7, ESI†) whereas the product synthesized without BCn shows very low BET surface area (Fig. S6, ESI†), again indicating that the presence of both BCn and APS is a vital requirement to achieve highly nanoporous silicas with pore apertures near the boundary between micro- and mesopores (vide supra).
Due to the non-covalent bonding of BCn and APS, removal of BCn by extraction was also investigated to potentially produce nanoporous silica with amine functional groups,11 demonstrated here for the silica synthesized at x = 0.18 with BC6. After three times of extraction, the removal of BC6 was evidenced by 1H–13C CP/MAS NMR and UV-Vis spectroscopies (Fig. S4 and S5, respectively, ESI†). As shown in Fig. S4 (ESI†), only resonance signals corresponding to APS are observed, suggesting that BC6 can be removed, while the APS moiety is still intact with the silica frameworks. This was further confirmed by 29Si MAS NMR, showing that the (Q2 + Q3 + Q4)/T3 peak ratio of the extracted sample is similar to that of the as-made one (Fig. S3, ESI†). It should be noted that in comparison with the as-made sample the peak area of Q3 silicon with respect to Q4 was slightly decreased upon both extraction and calcination, suggesting that some parts of silanol groups on/in the silica wall are condensed to form new Q4 silicon. The extracted sample exhibited a BET surface area of 730 m2 g−1, a micropore volume of 0.06 cm3 g−1, and a total pore volume of 0.35 cm3 g−1 (Fig. S12, ESI†). The reduction in porosity is partly due to the incomplete extraction of BC6 (about 20% remain), as suggested by the CHN elemental analyses (Table S1, ESI†). Further improvement of extraction conditions should be studied in the future. Note that the nitrogen content in the extracted sample is about 2 mmol g−1. The presence of such amine groups in the extracted samples can be useful for several applications, for example, in base catalysis, CO2 adsorption, and drug delivery.1
In summary, nanoporous silicas having some periodicity, high surface area, and pore diameters near the micro–meso transition region are synthesized using novel SDAs with aromatic cores and anionic end-groups. Their BET surface areas are among the highest values reported for nanoporous silicas. The pore diameters can be altered across the boundary between micro- and mesopores, ranging from 1.5 to 3 nm, depending on the size of SDAs (BCn) and the amount of co-SDA (APS). A possible formation scheme can be explained based on multiple, orthogonal interactions between SDAs, co-SDAs and silica frameworks. Upon dissolution in water, co-SDAs are deprotonated and they electrostatically interact with SDAs. Meanwhile, SDAs interact with each other via π–π interactions. Finally, the co-SDA and silica source co-condense to form the silica frameworks. We believe that although more studies are needed to understand the structure-directing mechanisms, this orthogonal approach can be extended to synthesize nanoporous silica with more complex functionality, which merits future investigation.
This work was supported in part by Grant-in-Aids for Scientific Research (A) (Grant Number: 26249118) and for Research Activity Start-up (Grant Number: 26889022) by the Japan Society for the Promotion of Science (JSPS). A part of this work was conducted at the Center for Nano Lithography & Analysis, The University of Tokyo, supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT). K. M. thanks the Program for Leading Graduate Schools, “Global Leader Program for Social Design and Management (GSDM),” by MEXT for financial support.
Notes and references
- (a) M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed; (b) K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1 CrossRef CAS; (c) K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26, 211 CrossRef CAS; (d) Y. Yamauchi, J. Ceram. Soc. Jpn., 2013, 121, 831 CrossRef CAS; (e) N. L. Torad, M. Naito, J. Tatami, A. Endo, S.-Y. Leo, S. Ishihara, K. C.-W. Wu, T. Wakihara and Y. Yamauchi, Chem. – Asian J., 2014, 9, 759 CrossRef CAS PubMed.
- (a) J. Jiang, J. L. Jorda, J. Yu, L. A. Baumes, E. Mugnaioli, M. J. Diaz-Cabanas, U. Kolb and A. Corma, Science, 2011, 333, 1131 CrossRef CAS PubMed; (b) J. Sun, C. Bonneau, Á. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154 CrossRef CAS PubMed; (c) L. Zhang, Q. Yang, H. Yang, J. Liu, H. Xin, B. Mezari, P. C. M. M. Magusin, H. C. L. Abbenhuis, R. A. van Santen and C. Li, J. Mater. Chem., 2008, 18, 450 RSC.
- (a) R. F. Lobo, Nature, 2006, 443, 757 CrossRef CAS PubMed; (b) J. Rouquerol, F. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Academic Press, Oxford, 2nd edn, 2014 Search PubMed.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120 CrossRef CAS PubMed.
- (a) A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, C. Martínez and M. Moliner, Nature, 2006, 443, 842 CrossRef CAS PubMed; (b) J. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 4986 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- (a) S.-L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950 RSC; (b) E. Elacqua, D. S. Lye and M. Weck, Acc. Chem. Res., 2014, 47, 2405 CrossRef CAS PubMed; (c) R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed; (d) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- (a) C. Li, Ö. Dag, T. D. Dao, T. Nagao, Y. Sakamoto, T. Kimura, O. Terasaki and Y. Yamauchi, Nat. Commun., 2015, 6, 6608 CrossRef CAS PubMed; (b) H. Wang, S. Ishihara, K. Ariga and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 10819 CrossRef CAS PubMed.
- (a) M. Kimura, K. Wada, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, J. Am. Chem. Soc., 2001, 123, 2438 CrossRef CAS; (b) A. Okabe, T. Fukushima, K. Ariga and T. Aida, Angew. Chem., Int. Ed., 2002, 41, 3414 CrossRef CAS; (c) H. O. Lintang, K. Kinbara, K. Tanaka, T. Yamashita and T. Aida, Angew. Chem., Int. Ed., 2010, 49, 4241 CrossRef CAS PubMed.
- Y. Fukasawa, A. Sugawara, H. Hirahara, A. Shimojima and T. Okubo, Chem. Lett., 2010, 39, 236 CrossRef CAS.
- (a) T. Yokoi, H. Yoshitake and T. Tatsumi, Chem. Mater., 2003, 15, 4536 CrossRef CAS; (b) S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801 CrossRef CAS PubMed; (c) T. Yokoi, H. Yoshitake, T. Yamada, Y. Kubota and T. Tatsumi, J. Mater. Chem., 2006, 16, 1125 RSC; (d) H. Zheng, C. Gao and S. Che, Microporous Mesoporous Mater., 2008, 116, 299 CrossRef CAS PubMed; (e) L. Han and S. Che, Chem. Soc. Rev., 2013, 42, 3740 RSC.
- (a) N. A. Brunelli, K. Venkatasubbaiah and C. W. Jones, Chem. Mater., 2012, 24, 2433 CrossRef CAS; (b) N. A. Brunelli and C. W. Jones, J. Catal., 2013, 308, 60 CrossRef CAS PubMed.
- (a) D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154 CrossRef PubMed; (b) L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287 RSC; (c) A. Corma, M. J. Díaz-Cabañas, M. Moliner and G. Rodríguez, Chem. Commun., 2006, 3137 RSC.
- (a) D. Xu, Y. Ma, Z. Jing, L. Han, B. Singh, J. Feng, X. Shen, F. Cao, P. Oleynikov, H. Sun, O. Terasaki and S. Che, Nat. Commun., 2014, 5, 4262 CAS; (b) B. K. Singh, D. Xu, L. Han, J. Ding, Y. Wang and S. Che, Chem. Mater., 2014, 26, 7183 CrossRef CAS.
- W. Chaikittisilp, A. Sugawara, A. Shimojima and T. Okubo, Chem. Mater., 2010, 22, 4841 CrossRef CAS.
- M. Thommes and K. A. Cychosz, Adsorption, 2014, 20, 233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization results of SDAs, NMR and UV-Vis spectra, FESEM and HRTEM images, nitrogen and argon sorption isotherms and their corresponding NL-DFT pore size distributions, and XRD patterns. See DOI: 10.1039/c5cc02801a |
| This journal is © The Royal Society of Chemistry 2015 |