
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the formation of discrete coordination nanostructures†
Joerg
Meyer
a,
Anja
Nickel
a,
Robin
Ohmann‡
a,
Lokamani
a,
Cormac
Toher§
a,
Dmitry A.
Ryndyk
a,
Yves
Garmshausen
b,
Stefan
Hecht
b,
Francesca
Moresco
*a and
Gianaurelio
Cuniberti
ac
aInstitute for Materials Science, Max Bergmann Center of Biomaterials, and Center for Advancing Electronics Dresden, TU Dresden, 01062 Dresden, Germany. E-mail: francesca.moresco@tu-dresden.de
bDepartment of Chemistry & IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
cDresden Center for Computational Materials Science (DCCMS), TU Dresden, 01062 Dresden, Germany
First published on 6th July 2015
Abstract
Novel surface coordination nanostructures based on cyanosexiphenyl molecules are assembled on a gold surface and investigated by scanning tunneling microscopy and density functional theory. Their formation can be tuned by varying the surface temperature during deposition. Diffusing gold adatoms act as coordination centers for the cyano groups present on one end of the nonsymmetrical molecules.
For the development of nanotechnology and atomic scale electronics, it is crucial to control the formation of single supramolecular structures at atomically clean surfaces with high precision. A central goal of research is therefore to fabricate atomically precise nanostructures with well-defined tailored properties. As many of the properties of supramolecular assemblies decisively depend on the nature of their intermolecular bonds, the precise tuning of the bond formation is, in this respect, of fundamental importance.1
Directed interactions, such as metal coordination, play a central role in building well-defined and extended nanostructures in a predictable and reproducible manner. In order to use metallic substrates to guide nanostructure formation, it is crucial to understand the interaction between the molecular building blocks and to control diffusion processes. Among the different non-covalent intermolecular bonds, metal coordination is especially important because it allows for the formation of structures with specific topologies and a high structural stability.1 Extended metal coordination networks on surfaces have been intensively studied in the last few years by scanning tunneling microscopy (STM) showing that ordered networks with a high binding energy can be formed on surfaces starting from a wide range of organic molecules and metal centers.2 However, less attention has been paid to the formation of discrete metal coordination complexes on surfaces, which can form stable non-covalent nanostructures with specific tailored properties.3
The formation of metal-coordination bonds requires both electron-donating ligands and metal centers. Such metal centers are either present on the metal surface under investigation,2c,g,4 or can be provided in a separate deposition step.2b Several examples have been recently reported where metal adatoms are used as coordination metal centers for molecules with cyano functional groups.2c,3e,5
Despite the recent interest in gold catalysis and the importance of gold coordination chemistry,6 coordination structures composed by cyano functional groups and gold adatoms were rarely investigated so far on a surface.7 Several other functional groups are known to strongly interact with gold atoms.8 In some cases,9 the gold atoms were assumed to be extracted by the molecules from the substrate. Self-assembly induced by cyano groups of porphyrins has been investigated,3a,10 observing CN–NC dipolar interactions on Au(111) and Cu(111). In such examples, the formation of CN–Cu–NC coordination bond was observed on Cu(111) after thermal annealing above 150 °C, whereas it was not observed on Ag(111) and Au(111). On a gold surface however, adatoms are known to diffuse on the surface at room temperature.11 Furthermore, a few examples of surface metal-coordination bond involving Au atoms were recently demonstrated.7,12
Here, we present the controlled fabrication of discrete coordination nanostructures formed by two or three molecules on a gold surface. For this goal, we specifically designed the cyanosexiphenyl molecule, schematically shown in Fig. 1a, which is an evaporable, nonsymmetrical, π-conjugated molecule.13 It consists of a sexiphenyl rod with a single terminal cyano group, which allows for selective linking of the sexiphenyl terminus to a gold atom via a Au–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C metal–ligand coordination bond.
C metal–ligand coordination bond.
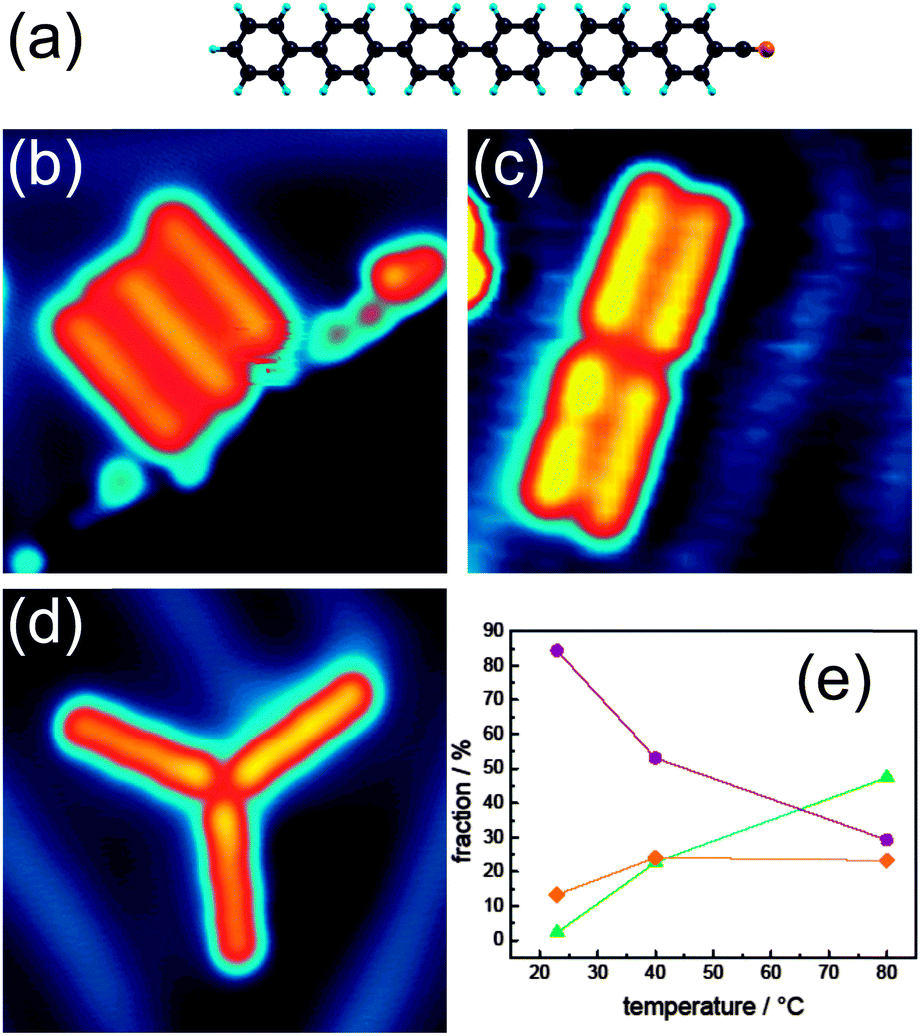 | ||
| Fig. 1 (a) Structural formula of cyanosexiphenyl (color code: nitrogen = orange, carbon = black, hydrogen = blue); (b)–(d) STM images of cyanosexiphenyl nanostructures on Au(111), (b) parallel structure at a step edge, (c) pair of dimers, (d) trimer (image parameters: V = −0.5 V, I = 0.2 nA, image size 7.5 nm × 7.5 nm); (e) fraction of cyanosexiphenyl nanostructures on Au(111) for different sample temperatures during preparation. Orange squares: molecules adsorbed on step edges and defects (b). Violet circles: dimer structures (c) and the green triangles represent the trimer structures (d). | ||
The investigation was performed by STM at low temperature (5 K) in UHV conditions, supported by density functional theory (DFT) calculations. Without adding any metal atoms and by varying the surface temperature, we can tune the formation of metal coordination structures of two or three molecules. By increasing the surface temperature during the deposition of the molecules, we can increase the diffusion of molecules and nanostructures on the surface. The nonsymmetrical molecules used in this work allow us to study defined, discrete nanostructures at submonolayer molecular coverage. This permits the investigation of metal–organic bonding in a case where, at monolayer coverage, the structure formation is dominated by hydrogen-bonding.3 In contrast to previous studies on self-assembled monolayers, the nanostructures can further diffuse and react on the surface, increasing the possibilities of molecular combination and opening a new possibility for the on-surface formation of specifically tailored functional nanostructures.
Cyanosexiphenyl molecules (Fig. 1a) are sublimated at 415 K onto Au(111) at submonolayer coverage and self-assemble into discrete supramolecular structures. As in the case of many other organic molecules, cyanosexiphenyl structures preferentially form at the fcc domains of the Au(111) surface reconstruction.14 At step edges, cyanosexiphenyl normally forms parallel arrangements of several molecules (Fig. 1b). No isolated molecules are visible on the substrate. Overview STM images are shown in Fig. S3 of the ESI.†
Structures composed of two molecules (dimers) are common when the surface is kept at room temperature (a pair of dimers is shown in Fig. 1c). Interestingly, these dimers form islands with other dimers and no single, isolated dimer can be observed on the surface. By increasing the temperature of the substrate, structures formed by three molecules (trimers) become dominant (Fig. 1d). The threefold symmetry of such structures reflects the symmetry of the Au(111) surface. The probability of assembling the different structures was experimentally investigated as a function of the substrate temperature during deposition (varying between 20 °C and 80 °C). The fraction of molecules forming the different nanostructures at different substrate temperatures during evaporation is given in Fig. 1e. More than 800 molecular structures were used for the statistics. As one can see, the fraction of molecules adsorbing at step edges and defects remains nearly constant with the substrate temperature in this range. However, the fraction of dimers decreases by increasing the temperature while the fraction of trimers increases. This suggests that the trimer structure is the most thermodynamically stable assembly and that a certain kinetic barrier has to be overcome to allow a dimer to accommodate a third molecule.
In the following, we derive a structural model of the observed nanostructures. The dimers form small islands in a parallel alignment, where the cyano groups are directly facing each other.
This is only possible if the two cyano groups have a common binding partner, which is a gold atom in the present case, and not by a CN–NC bonding resulting in an off-center arrangement.5a,b,15
Also for the trimers, the axes of the three molecules intersect in one point, suggesting again the presence of a common binding partner, as reported for similar systems in the literature.5b Structural models superposed to Fig. 1c and d are reported in the ESI† (Fig. S4 and S5). Such models show that a CN–NC bonding is geometrically not compatible with the observed structures.
Therefore, based on the molecular arrangement, we propose that both trimer and dimer structures are formed by coordination bonds between the cyano groups and gold adatoms. From the models reported in the ESI† we estimate a Au–N bonding length of about 2 Å. As already observed in several experiments, the metal atoms of metal coordination bonds are normally not resolved by STM.1a,2a–f,16
To confirm that the observed nanostructures are formed by metal coordination between gold adatoms and cyano functional groups, we performed DFT calculations, comparing the adsorption of a cyanosexiphenyl molecule on the bare surface with the adsorption at a gold adatom. When the molecule binds to an adatom, the configuration of the adsorbed molecule changes and the cyano group is raised up. The bonding energy of the molecule adsorbed at an adatom is calculated to be 6.64 eV. This is almost 0.4 eV larger than in the case without the Au adatom and confirms that the molecule preferentially adsorbs at gold adatoms on the surface (see ESI† for further details). Moreover, DFT calculations show a distance between Au and N in dimers of about 2.2 Å. As the structure is not flat, the projected distance on the (x,y) plane is about 1.6 Å. For a trimer, the calculated Au–N distance is slightly larger: 2.3 Å, and about 1.7 Å on the (x,y) plane. This is in good agreement with the value of 2 Å estimated from the model based on the experimental data (see ESI†).
We investigated the dependence of the nanostructure formation on the substrate temperature shown in Fig. 1e, calculating by DFT the diffusion barrier for different nanostructures on Au(111) (Fig. 2). To reduce the computational effort, molecules with only two phenyl rings (i.e. cyanobiphenyl) instead of six as in the case of cyanosexiphenyl are considered.
 | ||
| Fig. 2 Diffusion barrier height calculated for the different nanostructures on Au(111), considering cyanobiphenyl molecules. | ||
In order to determine the diffusion barriers of the different structures, we performed single-point energy calculations for the different possible adsorption sites. As one can see in Fig. 2, the diffusion barrier for the dimer pairs is the highest and about 0.3 eV larger than that of the lone dimers. This suggests that, once the dimer-pairs are formed from the lone dimers, which in turn are formed from the molecular structures of Fig. 3b and c, the dimer-pairs tend to stay at one place. The trimers (Fig. 3d) have a similar diffusion barrier to the dimer-pairs and are not mobile after formation, either. Since we omitted parts of the molecule to minimize computational efforts, the exact numbers given in Fig. 2 might be different for cyanosexiphenyl and we expect the diffusion barriers to be considerably higher due to the increased molecule–substrate interaction with the additional phenyl rings. Nevertheless, the qualitative differences between the molecules and their self-assembled structures will not be changed.
 | ||
| Fig. 3 Calculated nanostructures (cyanobiphenyl). The structure of the dimer-pair is shown in (a). A side view of a single molecule without adatom and a single molecule with adatom are presented in (b) and (c), respectively. The structure of the trimer is shown in (d). Note that for the calculations in this figure four phenyl rings are omitted to save computational effort. | ||
Increasing the substrate temperature induces flap dynamics in the mobile dimer with the adatom being the pivot-point. The energy barrier for this type of motion is low, so that the molecules in the dimer can easily rotate or “flap” around the adatom, allowing a third molecule to join and form a trimer. We investigated the formation of such trimers by calculating the binding energy for single cyanobiphenyl molecules on the surface diffusing towards the dimer with fixed pivot angle θ and found the approach to take place from the convex side as shown in the inset of Fig. 4.
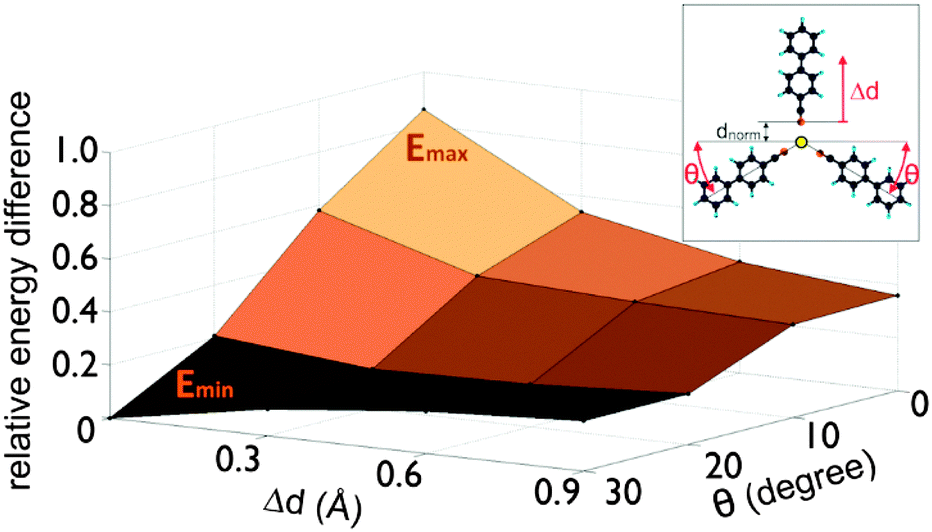 | ||
| Fig. 4 Binding energy of the trimer build from a dimer with fixed pivot angle θ and an approaching single molecule. The approaching way is shown schematically in the inset. The pivot angle θ and approached distance Δd are marked in red, while the normal binding distance dnorm is marked in black. The energy surface as a function of θ and Δd shows its minimum for the highest pivot angle and the lowest distance. | ||
The diffusion barrier for an approaching single cyanobiphenyl molecule is large for an unbent dimer but vanishes for the largest calculated convex angle of the bent dimer (see Fig. 4), suggesting that enhanced flap dynamics facilitate trimer formation by productive collisions of the third molecule with the bent dimer. In other words, simulations show that there are two competing processes important near room temperature: the formation of dimers and dimer-pairs on the one hand, and the formation of trimers from dimers and single molecules on the other.
At room temperature, two cyanosexiphenyl molecules tend to form a dimer-structure with a single gold adatom. These dimers are still mobile on the Au(111) surface. Depending on the substrate temperature, the dimers can arrange in a parallel fashion forming islands or build isolated trimer-structures by docking one additional cyanosexiphenyl molecule.
In conclusion, we have studied the formation of discrete metal coordination nanostructures on a Au(111) surface composed of two or three cyanosexiphenyl molecules. The nonsymmetrical molecules designed for this work allow for the fabrication of defined nanostructures at low coverage. The formation of the different nanostructures can be controlled by varying the substrate temperature during the evaporation of cyanosexiphenyl. DFT calculations confirm the formation of metal–ligand coordination bonds between the cyano groups and the Au adatoms.
Assemblies of a few molecules bonded by metal coordination bonds represent an alternative route for the fabrication of stable organic nanostructures with defined properties. This is essential to increase the reproducibility and diversity of surface-assembled nanomaterials and thereby improve bottom-up nanotechnology.
This work was supported by the ICT-FET European Union Integrated Project PAMS, the Helmholtz Association (via Helmholtz Energy Alliance and IHRS Nanonet VH-KO-606), the Deutsche Forschungsgemeinschaft via SFB 951, and the DFG and the National Science Foundation via the common project NSF 11-568. We gratefully acknowledge support from the German Excellence Initiative via the Cluster of Excellence EXC1056 “Center for Advancing Electronics Dresden” (cfaed). We thank Dr Michael Pätzel and Björn Kobin for help during the initial synthesis of cyanosexiphenyl. Computational facilities were provided by Zentrum für Informationsdienste und Hochleistungsrechnen (ZIH) at TU Dresden.
Notes and references
- (a) L. Bartels, Nat. Chem., 2010, 2, 87 CrossRef CAS PubMed; (b) S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139 RSC.
- (a) N. Lin, A. Dmitriev, J. Weckesser, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2002, 41, 4779 CrossRef CAS PubMed; (b) S. Stepanow, N. Lin, D. Payer, U. Schlickum, F. Klappenberger, G. Zoppellaro, M. Ruben, H. Brune, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 710 CrossRef CAS PubMed; (c) G. Pawin, K. L. Wong, D. Kim, D. Sun, L. Bartels, S. Hong, T. S. Rahman, R. Carp and M. Marsella, Angew. Chem., Int. Ed., 2008, 47, 8442 CrossRef CAS PubMed; (d) U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813 CrossRef CAS PubMed; (e) A. Langner, S. L. Tait, N. Lin, C. Rajadurai, M. Ruben and K. Kern, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17927 CrossRef CAS PubMed; (f) S. Jensen and C. J. Baddeley, J. Phys. Chem. C, 2008, 112, 15439 CrossRef CAS; (g) H. Walch, J. Dienstmaier, G. Eder, R. Gutzler, S. Schlögl, T. Sirtl, K. Das, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2011, 133, 7909 CrossRef CAS PubMed.
- (a) T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619 CrossRef CAS PubMed; (b) C. C. Perry, S. Haq, B. G. Frederick and N. V. Richardson, Surf. Sci., 1998, 409, 512 CrossRef CAS; (c) P. Messina, A. Dmitriev, N. Lin, H. Spillmann, M. Abel, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2002, 124, 14000 CrossRef CAS PubMed; (d) L.-A. Fendt, M. Stöhr, N. Wintjes, M. Enache, T. A. Jung and F. Diederich, Chem. – Eur. J., 2009, 15, 11139 CrossRef CAS PubMed; (e) N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138 CrossRef CAS PubMed; (f) L. Vitali, G. Levita, R. Ohmann, A. Comisso, A. De Vita and K. Kern, Nat. Mater., 2010, 9, 320 CrossRef CAS PubMed; (g) T. Lin, X. S. Shang, P. N. Liu and N. Lin, J. Phys. Chem. C, 2013, 117, 23027 CrossRef CAS.
- R. Ohmann, L. Vitali and K. Kern, Nano Lett., 2010, 10, 2995 CrossRef CAS PubMed.
- (a) T. Sirtl, S. Schlögl, A. Rastgoo-Lahrood, J. Jelic, S. Neogi, M. Schmittel, W. M. Heckl, K. Reuter and M. Lackinger, J. Am. Chem. Soc., 2013, 135, 691 CrossRef CAS PubMed; (b) M. Pivetta, G. E. Pacchioni, E. Fernandes and H. Brune, J. Chem. Phys., 2015, 142, 101928 CrossRef PubMed; (c) N. Henningsen, R. Rurali, C. Limbach, R. Drost, J. I. Pascual and K. J. Franke, J. Phys. Chem. Lett., 2011, 2, 55 CrossRef CAS; (d) D. Écija, J. I. Urgel, A. C. Papageorgiou, S. Joshi, W. Auwärter, A. P. Seitsonen, S. Klyatskaya, M. Ruben, S. Fischer, S. Vijayaraghavan, J. Reichert and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6678 CrossRef PubMed.
- A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC.
- Z. Yang, M. Corso, R. Robles, C. Lotze, R. Fitzner, E. Mena-Osteritz, P. Bäuerle, K. J. Franke and J. I. Pascual, ACS Nano, 2014, 8, 10715 CrossRef CAS PubMed.
- (a) P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 146103 CrossRef; (b) J. A. Boscoboinik, F. C. Calaza, Z. Habeeb, D. W. Bennett, D. J. Stacchiola, M. A. Purino and W. T. Tysoe, Phys. Chem. Chem. Phys., 2010, 12, 11624 RSC.
- F. Rossel, P. Brodard, F. Patthey, N. V. Richardson and W. D. Schneider, Surf. Sci., 2008, 602, L115 CrossRef CAS PubMed.
- (a) Y. Okuno, T. Yokoyama, S. Yokoyama, T. Kamikado and S. Mashiko, J. Am. Chem. Soc., 2002, 124, 7218 CrossRef CAS PubMed; (b) C. Iacovita, P. Fesser, S. Vijayaraghavan, M. Enache, M. Stöhr, F. Diederich and T. A. Jung, Chem. – Eur. J., 2012, 18, 14610 CrossRef CAS PubMed.
- R. Jaklevic and L. Elie, Phys. Rev. Lett., 1988, 60, 120 CrossRef CAS.
- (a) Y. Pan, B. Yang, C. Hulot, S. Blechert, N. Nilius and H.-J. Freund, Phys. Chem. Chem. Phys., 2012, 14, 10987 RSC; (b) Z. Shi and N. Lin, J. Am. Chem. Soc., 2009, 131, 5376 CrossRef CAS PubMed.
- Y. Garmshausen, J. Schwarz, J. Hildebrandt, B. Kobin, M. Pätzel and S. Hecht, Org. Lett., 2014, 16, 2838 CrossRef CAS PubMed.
- M. Böhringer, K. Morgenstern, W.-D. Schneider, R. Berndt, F. Mauri, A. De Vita and R. Car, Phys. Rev. Lett., 1999, 83, 324 CrossRef.
- D. Kühne, F. Klappenberger, R. Decker, U. Schlickum, H. Brune, S. Klyatskaya, M. Ruben and J. V. Barth, J. Phys. Chem. C, 2009, 113, 17851 Search PubMed.
- (a) A. Dmitriev, H. Spillmann, N. Lin, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2003, 42, 2670 CrossRef CAS PubMed; (b) A. Langner, S. L. Tait, N. Lin, R. Chandrasekar, M. Ruben and K. Kern, Angew. Chem., Int. Ed., 2008, 47, 8835 CrossRef CAS PubMed; (c) T. Classen, G. Fratesi, G. Costantini, S. Fabris, F. L. Stadler, C. Kim, S. de Gironcoli, S. Baroni and K. Kern, Angew. Chem., Int. Ed., 2005, 44, 6142 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc02723c |
| ‡ Present address: Okinawa Institute of Science and Technology (OIST), 1919-1 Tancha, Onna-son, Okinawa, 904-0495 Japan. |
| § Present address: Department of Mechanical Engineering and Materials Science, Duke University, Durham, NC 27708, USA. |
| This journal is © The Royal Society of Chemistry 2015 |