
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed α-selective hydrostannylation of alkynes for the synthesis of branched alkenylstannanes†
H.
Yoshida
*ab,
A.
Shinke
a,
Y.
Kawano
a and
K.
Takaki
a
aDepartment of Applied Chemistry, Graduate School of Engineering, Hiroshima University, Higashi-Hiroshima 739-8527, Japan. E-mail: yhiroto@hiroshima-u.ac.jp; Fax: +81-82-424-5494; Tel: +81-82-424-7724
bACT-C, Japan Science and Technology Agency, Higashi-Hiroshima 739-8527, Japan
First published on 27th April 2015
Abstract
A variety of branched alkenylstannanes can directly be synthesized with excellent α-selectivity by the copper-catalyzed hydrostannylation using a distannane or a silylstannane, irrespective of the electronic and steric characteristics of terminal alkynes employed. Synthetic utility of the resulting branched alkenylstannane has been demonstrated by the total synthesis of bexarotene.
In view of the high synthetic versatility of alkenylstannanes, especially in carbon–carbon bond-forming processes via Migita–Kosugi–Stille coupling, tin–lithium exchange reaction, etc., the development of potent methods for making alkenylstannanes with defined structures in a regio- and stereoselective manner has continued to be a key subject in modern synthetic organic chemistry.1 One of the most popular and straightforward routes to alkenylstannanes would be hydrostannylation of alkynes,2 and three isomers, namely the α-adduct and the (E/Z)-β-adduct, can be generated in the case of terminal alkynes (eqn (1)).3 Hence the regio- and stereocontrol of hydrostannylation is a pivotal issue, and (Z)- or (E)-linear alkenylstannanes have successfully been synthesized with high β-selectivity under radical conditions,4 Lewis acid5 or transition metal catalysis.6 Although selective access to branched alkenylstannanes has also been achieved in some cases depending upon direct addition of a tin hydride7–9 or a stannylmetallation–protonation sequence,10 the existing methods are still not versatile, owing to limited substrate scope of alkynes in every reaction and the use of an excess amount of a stannylmetal reagent in the latter case (Scheme 1). Therefore the development of a universal system for α-selective hydrostannylation of terminal alkynes, which gives direct and efficacious access to pharmacologically important 1,1-disubstituted alkenes such as bexarotene11 and isocombretastatin A-4,12 has been a long-awaited goal.
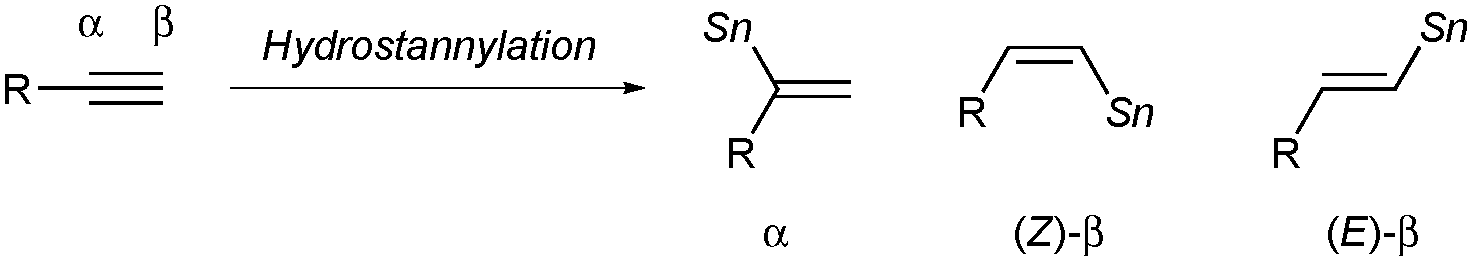 | (1) |
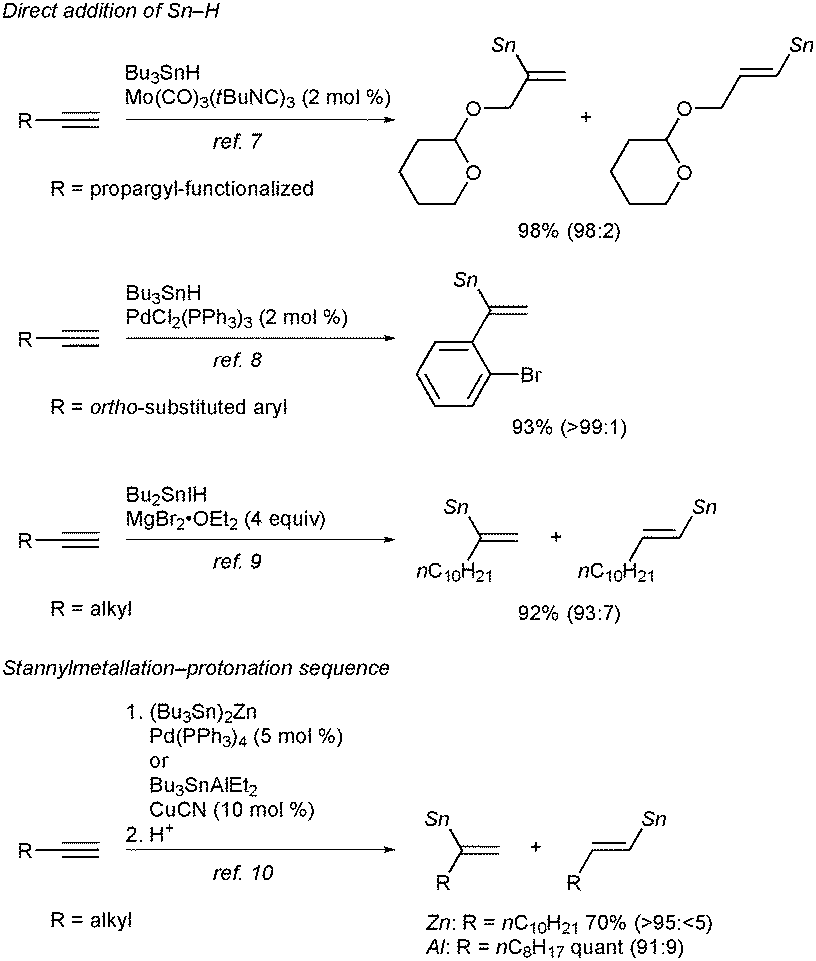 | ||
| Scheme 1 Reported α-selective hydrostannylation of terminal alkynes. | ||
We have recently riveted our attention on potential copper catalysis for metallation reactions of unsaturated carbon–carbon bonds, and have already disclosed that distannylation13 as well as various borylations14 of alkynes and alkenes facilely occur to afford organostannanes and -boranes with structural diversity by employing a distannane and a diboron as a metallating reagent. The distannylation of alkynes was found to proceed through intermediary formation of a β-stannylalkenylcopper species with enough nucleophilicity, which was finally converted into vic-distannylalkenes by capturing a tin electrophile. Therefore, we envisioned that a copper catalyst would also promote hydrostannylation of alkynes in the presence of a suitable protic reagent for trapping the β-stannylalkenylcopper intermediate.15 Herein we report that the hydrostannylation of terminal alkynes smoothly takes place under the copper catalysis by use of water as a protic reagent, and that the universal system allows a variety of branched alkenylstannanes to be synthesized with excellent α-selectivity, irrespective of the electronic and steric characteristics of terminal alkynes.
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2′a = 98:2), when we treated 1-tetradecyne (1a) with hexamethyldistannane and water in toluene at 110 °C in the presence of the Cu(OAc)2–PtBu3 catalyst (Table 1, entry 1).16,17 The reaction was also applicable to 1-dodecyne (1b) and an imide-substituted alkyne (1c), giving 2b and 2c with high degrees of α-selectivity in excellent yield (entries 2 and 3), and furthermore functionalized aliphatic alkynes bearing an acetal (1d), a silyl ether (1e) or a benzyl ether (1f) smoothly underwent the α-selective hydrostannylation, leaving these reactive moieties intact (entries 4–6). The high functional group compatibility was also demonstrated by the reaction of a hydroxyl- (1g) or a bromo-substituted alkyne (1h), and propargyl-functionalized alkynes (1i and 1j), although the yields became moderate in some cases (entries 7–10). The regioselective installation of a stannyl group into an internal carbon of aromatic terminal alkynes was achieved under the present conditions as well, and thus pyridyl (1k), naphthyl (1l) and phenyl (1m and 1n) acetylenes were efficiently transformed into the respective branched alkenylstannanes (2k–2n) (entries 11–14). An internal alkyne, diphenylacetylene (1o), could participate in the reaction to furnish (E)-trimethylstannylstilbene (2o) as the sole product (eqn (2)), showing that hydrostannylation completely proceeds in a cis fashion.18
:2′a = 98:2), when we treated 1-tetradecyne (1a) with hexamethyldistannane and water in toluene at 110 °C in the presence of the Cu(OAc)2–PtBu3 catalyst (Table 1, entry 1).16,17 The reaction was also applicable to 1-dodecyne (1b) and an imide-substituted alkyne (1c), giving 2b and 2c with high degrees of α-selectivity in excellent yield (entries 2 and 3), and furthermore functionalized aliphatic alkynes bearing an acetal (1d), a silyl ether (1e) or a benzyl ether (1f) smoothly underwent the α-selective hydrostannylation, leaving these reactive moieties intact (entries 4–6). The high functional group compatibility was also demonstrated by the reaction of a hydroxyl- (1g) or a bromo-substituted alkyne (1h), and propargyl-functionalized alkynes (1i and 1j), although the yields became moderate in some cases (entries 7–10). The regioselective installation of a stannyl group into an internal carbon of aromatic terminal alkynes was achieved under the present conditions as well, and thus pyridyl (1k), naphthyl (1l) and phenyl (1m and 1n) acetylenes were efficiently transformed into the respective branched alkenylstannanes (2k–2n) (entries 11–14). An internal alkyne, diphenylacetylene (1o), could participate in the reaction to furnish (E)-trimethylstannylstilbene (2o) as the sole product (eqn (2)), showing that hydrostannylation completely proceeds in a cis fashion.18
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time (h) | Yieldb (%) |
2:2′ |
Products |
| a General procedure: 1 (0.30 mmol, 1 equiv.), Me3Sn–SnMe3 (0.39 mmol, 1.3 equiv.), H2O (0.90 mmol, 3 equiv.), Cu(OAc)2 (0.015 μmol, 5 mol%), PtBu3 (0.053 mmol, 17.5 mol%), toluene (0.2 mL). b Isolated yield. | |||||
| 1 | nC12H25 (1a) | 1.5 | 75 | 98:2 |
2a,2′a |
| 2 | nC10H21 (1b) | 2 | 89 | 97:3 |
2b,2′b |
| 3 |
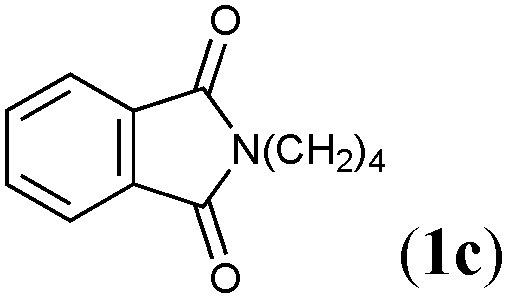
|
1.5 | 89 | 97:3 |
2c,2′c |
| 4 | THPO(CH2)4 (1d) | 1 | 84 | 94:6 |
2d,2′d |
| 5 | TBSO(CH2)2 (1e) | 1.5 | 85 | 96:4 |
2e,2′e |
| 6 | BnO(CH2)2 (1f) | 1 | 76 | 97:3 |
2f,2′f |
| 7 | HO(CH2)8 (1g) | 3 | 76 | >99:1 |
2g |
| 8 | Br(CH2)8 (1h) | 24 | 43 | 89:11 |
2h,2′h |
| 9 |

|
8 | 63 | >99:1 |
2i |
| 10 | BnOCH2 (1j) | 6 | 37 | 92:8 |
2j,2′j |
| 11 | 2-Pyridyl (1k) | 5.5 | 68 | >99:1 |
2k |
| 12 |

|
6 | 68 | 86:14 |
2l,2′l |
| 13 | 4-nBuC6H4 (1m) | 24 | 61 | >99:1 |
2m |
| 14 |

|
2 | 80 | >99:1 |
2n |

With the successful synthesis of diverse branched alkenylstannanes having a trimethylstannyl moiety, we next investigated α-selective installation of a tributylstannyl moiety. Although the reaction of 1-octyne (1p) with hexabutyldistannane in the presence of the Cu(OAc)2–PCy3 catalyst19 led to regioselective formation of a branched alkenylstannane (3p) in moderate yield (Table 2, entry 1), a silylstannane, tributyl(trimethylsilyl)stannane, turned out to serve as a more effective and reactive stannylating reagent to afford a 74% yield of 3p and 3′p (3p:3′p = 86:14) (entry 2).17 It should be noted that a silyl moiety was not incorporated into an alkyne at all in the reaction with a silylstannane, which is in marked contrast to the copper-catalyzed selective silyl incorporation reactions into unsaturated hydrocarbons with a silylborane.20 Hydrostannylation using a silylstannane also took place smoothly with 1-decyne (1q), 1-hexyne (1r) and branched aliphatic terminal alkynes (1s and 1t) to provide 3q–3t with high α-selectivity (entries 3–6), and furthermore functionalized terminal alkynes bearing a cyano (1u), a chloro (1v) or a benzyloxy moiety (1f), and 3-phenyl-1-propyne (1w) were transformed into the respective branched alkenylstannanes (entries 7–10).21
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time (h) | Yieldb (%) |
3:3′ |
Products |
| a General procedure: 1 (0.45 mmol, 1.5 equiv.), Me3Si–SnBu3 (0.30 mmol, 1 equiv.), H2O (0.45 mmol, 1.5 equiv.), Cu(OAc)2 (0.03 μmol, 10 mol%), PCy3 (0.11 mmol, 35 mol%), toluene (0.2 mL). b Isolated yield. c Bu3Sn–SnBu3 was used instead of Me3Si–SnBu3. H2O = 0.4 mL (74 equiv.). d NMR yield. | |||||
| 1c | nC6H13 (1p) | 43 | 43d | >99:1 |
3p |
| 2 | nC6H13 (1p) | 4 | 73 | 86:14 |
3p,3′p |
| 3 | nC8H17 (1q) | 2 | 74 | 88:12 |
3q,3′q |
| 4 | nBu (1r) | 2 | 74 | 85:15 |
3r,3′r |
| 5 | iAmyl (1s) | 2 | 60 | 84:16 |
3s,3′s |
| 6 | iBu (1t) | 2 | 48 | 92:8 |
3t,3′t |
| 7 | NC(CH2)3 (1u) | 3 | 55 | 93:7 |
3u,3′u |
| 8 | Cl(CH2)3 (1v) | 2 | 44 | 92:8 |
3v,3′v |
| 9 | BnO(CH2)2 (1f) | 2 | 55 | 88:12 |
3f,3′f |
| 10 | Bn (1w) | 2 | 46 | 93:7 |
3w,3′w |

Since water can serve as a proton source in hydrostannylation, we expected that the present system may be extended to deuteriostannylation by use of deuterium oxide. Surprisingly, the copper-catalyzed reaction of 1-tetradecyne (1a) with hexamethyldistannane in the presence of deuterium oxide produced a dideuteriostannylation product (2a-d2) predominantly (2a-d2:2a-d:2a = 79:7:14, eqn (a), Scheme 2). The formation of 2a-d2 can be rationalized by considering the deuteriostannylation of 1-tetradecyne-d (1a-d), which should be generated in situ prior to deuteriostannylation. Actually, hydrogen–deuterium exchange between 1a and deuterium oxide was demonstrated to occur smoothly under the copper catalysis (eqn (b), Scheme 2).
 | ||
| Scheme 2 Cu-catalyzed deuteriostannylation with deuterium oxide. | ||
As depicted in Scheme 3, the branched alkenylstannane (2n) was found to be facilely converted into 1,1-diarylalkene 4 by the Migita–Kosugi–Stille coupling with ethyl 4-iodobenzoate. Hydrolysis of the ester moiety of 4 provided bexarotene 5 in 39% overall yield (3 steps, based on alkyne 1n), which is widely used in the treatment of cutaneous T-cell lymphoma,11 demonstrating the synthetic significance of the present α-selective hydrostannylation.
 | ||
| Scheme 3 Total synthesis of bexarotene. | ||
Formation of a stannylcopper species (6) via σ-bond metathesis between a distannane (or silylstannane) and Cu–OH would initiate hydrostannylation (Scheme 4).22 The resulting stannylcopper species (6) then adds across a carbon–carbon triple bond of a terminal alkyne (stannylcupration) to produce a β-stannylalkenylcopper species (7), which is finally transformed into a hydrostannylation product through protonation with water.23 The formation of branched alkenylstannanes (2 and 3) with high α-selectivity should be attributed to the regioselective generation of 7, possessing the stannyl moiety at the internal carbon, in the stannylcupration step, which has already been well documented to be kinetically favored in a stoichiometric reaction of a stannylcopper species with a terminal alkyne.24
 | ||
| Scheme 4 A plausible catalytic cycle for hydrostannylation. | ||
In conclusion, we have developed a universal system for the α-selective hydrostannylation of terminal alkynes by using a distannane or a silylstannane as a stannylating reagent in the presence of a copper–trialkylphosphine catalyst, which leads to a convenient and straightforward method for synthesizing diverse branched alkenylstannanes, irrespective of the electronic and steric nature of terminal alkynes employed. The resulting branched alkenylstannane has been demonstrated to be facilely transformed into bexarotene of pharmacologically importance via the cross-coupling reaction. Further studies on copper-catalyzed stannylation reactions using a distannane or a silylstannane are in progress.
Notes and references
- (a) M. Pereyre, J. P. Quintard and A. Rahm, Tin in Organic Synthesis, Butterworth, London, 1987 Search PubMed; (b) V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1 CAS; (c) A. Orita and J. Otera, in Tin in Organic Synthesis, Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley-VCH, Weinheim, 2004, pp. 621–720 Search PubMed; (d) T. N. Mitchell, Organotin Reagents in Cross-Coupling Reactions, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, pp. 125–161 Search PubMed; (e) A. G. Davies, Organotin Chemistry, Wiley-VCH, Weinheim, 2004 CrossRef PubMed.
- (a) N. D. Smith, J. Mancuso and M. Lautens, Chem. Rev., 2000, 100, 3257 CrossRef CAS PubMed; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 853 CrossRef CAS PubMed.
- For leading references, see: (a) Y. Ichinose, H. Oda, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1987, 60, 3468 CrossRef CAS; (b) K. Kikukawa, H. Umekawa, F. Wada and T. Matsuda, Chem. Lett., 1988, 881 CrossRef CAS; (c) H. X. Zhang, F. Guibé and G. Balavoine, J. Org. Chem., 1990, 55, 1857 CrossRef CAS.
- (a) E. Nakamura, D. Machii and T. Inubushi, J. Am. Chem. Soc., 1989, 111, 6849 CrossRef CAS; (b) E. Nakamura, Y. Imanishi and D. Machii, J. Org. Chem., 1994, 59, 8178 CrossRef CAS; (c) K. Miura, D. Wang, Y. Matsumoto, N. Fujisawa and A. Hosomi, J. Org. Chem., 2003, 68, 8730 CrossRef CAS PubMed; (d) K. Miura, D. Wang, Y. Matsumoto and A. Hosomi, Org. Lett., 2005, 7, 503 CrossRef CAS PubMed.
- (a) N. Asao, J.-X. Liu, T. Sudoh and Y. Yamamoto, J. Chem. Soc., Chem. Commun., 1995, 2405 RSC; (b) N. Asao, J.-X. Liu, T. Sudoh and Y. Yamamoto, J. Org. Chem., 1996, 61, 4568 CrossRef CAS PubMed; (c) V. Gevorgyan, J.-X. Liu and Y. Yamamoto, Chem. Commun., 1998, 37 RSC.
- (a) R. E. Maleczka Jr., L. R. Terrell, D. H. Clark, S. L. Whitehead, W. P. Gallagher and I. Terstiege, J. Org. Chem., 1999, 64, 5958 CrossRef; (b) A. Darwish, A. Lang, T. Kim and J. M. Chong, Org. Lett., 2008, 10, 861 CrossRef CAS PubMed . See also ref. 3c.
- (a) U. Kazmaier, D. Schauss and M. Pohlman, Org. Lett., 1999, 1, 1017 CrossRef CAS; (b) U. Kazmaier, M. Pohlman and D. Schauß, Eur. J. Org. Chem., 2000, 2761 CrossRef CAS; (c) S. Braune and U. Kazmaier, J. Organomet. Chem., 2002, 641, 26 CrossRef CAS; (d) S. Braune, M. Pohlman and U. Kazmaier, J. Org. Chem., 2004, 69, 468 CrossRef CAS PubMed; (e) H. Lin and U. Kazmaier, Eur. J. Org. Chem., 2007, 2839 CrossRef CAS PubMed; (f) N. Jena and U. Kazmaier, Eur. J. Org. Chem., 2008, 3852 CrossRef CAS PubMed; (g) H. Lin and U. Kazmaier, Eur. J. Org. Chem., 2009, 1221 CrossRef CAS PubMed.
- A. Hamze, D. Veau, O. Provot, J.-D. Brion and M. Alami, J. Org. Chem., 2009, 74, 1337 CrossRef CAS PubMed.
- I. Shibata, T. Suwa, K. Ryu and A. Baba, J. Am. Chem. Soc., 2001, 123, 4101 CrossRef CAS.
- (a) J. Hibino, S. Matsubara, Y. Morizawa, K. Oshima and H. Nozaki, Tetrahedron Lett., 1984, 25, 2151 CrossRef CAS; (b) S. Matsubara, J. Hibino, Y. Morizawa, K. Oshima and H. Nozaki, J. Organomet. Chem., 1985, 285, 163 CrossRef CAS; (c) S. Sharma and A. C. Oehlschlager, Tetrahedron Lett., 1986, 27, 6161 CrossRef CAS; (d) S. Sharma and A. C. Oehlschlager, Tetrahedron Lett., 1988, 29, 261 CrossRef CAS; (e) S. Sharma and A. C. Oehlschlager, J. Org. Chem., 1989, 54, 5064 CrossRef CAS.
- T. Illidge, C. Chan, N. Counsell, S. Morris, J. Scarisbrick, D. Gilson, B. Popova, P. Patrick, P. Smith, S. Whittaker and R. Cowan, Br. J. Cancer, 2013, 109, 2566 CrossRef CAS PubMed.
- J. Aziz, E. Brachet, A. Hamze, J.-F. Peyrat, G. Bernadat, E. Morvan, J. Bignon, J. Wdzieczak-Bakala, D. Desravines, J. Dubois, M. Tueni, A. Yassine, J.-D. Brion and M. Alami, Org. Biomol. Chem., 2013, 11, 430 CAS.
- H. Yoshida, A. Shinke and K. Takaki, Chem. Commun., 2013, 49, 11671 RSC.
- (a) H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2012, 51, 235 CrossRef CAS PubMed; (b) Y. Takemoto, H. Yoshida and K. Takaki, Chem. – Eur. J., 2012, 18, 14841 CrossRef CAS PubMed; (c) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2013, 15, 952 CrossRef CAS PubMed; (d) I. Kageyuki, H. Yoshida and K. Takaki, Synthesis, 2014, 1924 Search PubMed; (e) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2014, 50, 8299 RSC; (f) Y. Takemoto, H. Yoshida and K. Takaki, Synthesis, 2014, 3024 CAS; (g) H. Yoshida, Y. Takemoto and K. Takaki, Asian J. Org. Chem., 2014, 3, 1204 CrossRef CAS PubMed; (h) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2015, 51, 6297 RSC.
- For stoichiometric formation of a β-stannylalkenylcopper species, followed by its capture with an electrophile including protons, see: (a) A. Barbero, P. Cuadrado, I. Fleming, A. M. González and F. J. Pulido, J. Chem. Soc., Chem. Commun., 1992, 351 RSC; (b) A. Barbero, P. Cuadrado, I. Fleming, A. M. González, F. J. Pulido and R. Rubio, J. Chem. Soc., Perkin Trans. 1, 1993, 1657 RSC.
- B. Hammond, F. H. Jardine and A. G. Vohra, J. Inorg. Nucl. Chem., 1971, 33, 1017 CrossRef CAS.
- For optimization of reaction conditions (ligand, protic reagent, solvent, etc.), see ESI†.
- The reaction of 1-phenyl-1-butyne did not give a hydorostannylation product.
- The use of Cu(I) sources such as CuCl (with NaOtBu) and Cu(I)-thiophene-2-carboxylate resulted in lower yield.
- For representative examples, see: (a) A. H. Hoveyda and K.-S. Lee, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef PubMed; (b) D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513 CrossRef CAS PubMed; (c) P. Wang, X.-L. Yeo and T.-P. Loh, J. Am. Chem. Soc., 2011, 133, 1254 CrossRef CAS PubMed; (d) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487 CrossRef CAS PubMed.
- The relatively low yields with TMSSnBu3 compared with those with Me3Sn–SnMe3 are ascribed to the formation of Bu3Sn–SnBu3 as a by-product, which has been demonstrated to occur only in the presence of a Cu catalyst and a proton source. See ESI† for details.
- For generation of a stannylcopper species by use of a distannane or a silylstannane, see: (a) A. C. Oehlschlager, M. W. Hutzinger, R. Aksela, S. Sharma and S. M. Singh, Tetrahedron Lett., 1990, 31, 165 CrossRef CAS; (b) B. H. Lipshutz, D. C. Reuter and E. L. Ellsworth, J. Org. Chem., 1989, 54, 4975 CrossRef CAS.
- Addition of a stannylcopper species to an alkyne and subsequent protonation of the resulting β-stannylalkenylcopper species have been established steps in the stoichiometric stannylcupration of alkynes. See: (a) A. Barbero and F. J. Pulido, Chem. Soc. Rev., 2005, 34, 913 RSC; (b) F. J. Pulido and A. Barbero, Silyl and Stannyl Derivatives of Organocopper Compounds, in The Chemistry of Organocopper Compounds, ed. Z. Rappoport and I. Marek, John Wiley & Sons, Chichester, 2009, pp. 775–856 Search PubMed.
- R. D. Singer, M. W. Hutzzinger and A. C. Oehlschlager, J. Org. Chem., 1991, 56, 4933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: 10.1039/c5cc02720a |
| This journal is © The Royal Society of Chemistry 2015 |