
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect accessibility of mixed-metal (III/II) acid sites through the rational synthesis of porous metal carboxylates†
S.
Wongsakulphasatch
ab,
F.
Nouar
*a,
J.
Rodriguez
c,
L.
Scott
a,
C.
Le Guillouzer
d,
T.
Devic
a,
P.
Horcajada
a,
J.-M.
Grenèche
e,
P. L.
Llewellyn
c,
A.
Vimont
d,
G.
Clet
d,
M.
Daturi
d and
C.
Serre
*a
aInstitut Lavoisier (CNRS UMR 8180), Université de Versailles St-Quentin en Yvelines, 45 avenue des Etats-Unis, 78035 Versailles Cedex, France. E-mail: farid.nouar@uvsq.fr; christian.serre@uvsq.fr
bDepartment of Chemical Engineering, Faculty of Engineering and Industrial Technology Silpakorn University, Nakhon Pathom 73000, Thailand
cAix-Marseille Univ., CNRS, Laboratoire MADIREL (UMR 7246), Campus de Saint Jérôme, 13397, Marseille Cedex 20, France
dLaboratoire Catalyse et Spectrochimie, ENSICAEN, Université de Caen Basse-Normandie, CNRS, 6 Bd Maréchal Juin, 14050 Caen, France
eInstitut des Molécules et des Matériaux du Mans (CNRS UMR 6283), Université du Maine, 72085 Le Mans, France
First published on 14th May 2015
Abstract
The scalable and environmentally-friendly synthesis of mixed Fe(III)/M(II) (M = Ni, Co, Mg) polycarboxylate porous MOFs based on the Secondary Building Unit approach is reported. A combination of in situ infrared spectroscopy, 57Fe Mössbauer spectrometry and adsorption microcalorimetry confirms the direct accessibility of the iron(III) and metal(II) sites under low temperature activation conditions.
Due to their chemical and structural diversity, Metal–Organic Frameworks (MOFs) or Porous Coordination Polymers (PCPs) are an attractive class of porous crystalline materials. They have been evaluated for a wide range of potential applications, ranging from gas storage, separation, catalysis, sensing to biomedicine.1 Among the thousands of architectures reported so far, only a few of them are considered suitable for practical applications.2 It is indeed challenging to combine high porosity with sufficient chemical stability, scalability of the synthesis and desired chemical functionality. Iron(III) polycarboxylate MOFs are particularly attractive candidates due to their robustness, their high biocompatibility and their redox behaviour. Their potential interest in separation,3 catalysis4 or drug delivery5 has recently been confirmed. Typical examples are the series of highly flexible MIL-536 or MIL-88,7 the highly porous MIL-100,8 MIL-1019, PCN-53,10 MIL-1274a (or soc-MOF) and a BTB based solid with the topology11 or the porphyrin based iron MOFs MIL-14112 or PCN-600.13 Their functionalization (direct or via post-synthetic grafting) or the isoreticular concept allows a careful tuning of their structural or chemical features. Few of these solids4a,7–13 are built up from oxo-centered trimers of iron(III) octahedra (Fig. 1, left), which act as accessible metallic Fe(III) sites upon solvent departure. It is noteworthy that at a rather high activation temperature (T > 473 K) under vacuum or inert gas flow, it is possible to partially and reversibly reduce such sites. This leads to mixed valence iron(III/II) MOFs, the concomitant presence of Fe(III) and Fe(II) Lewis acid sites resulting in enhanced properties in separation, biomedicine or catalysis.3b
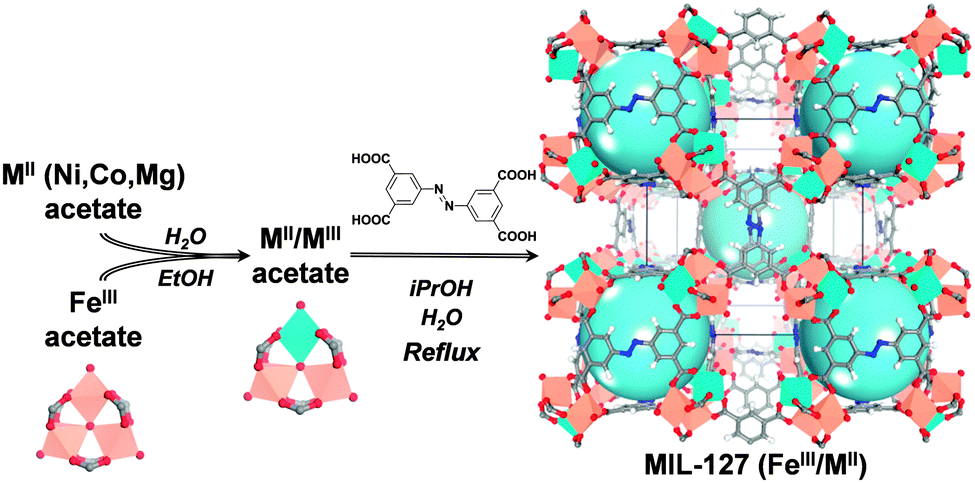 | ||
| Fig. 1 Schematic representation of the building block approach to produce mixed-metal carboxylate MOFs such as MIL-127. From left to right, mixing iron acetate trimeric building blocks with MII acetate compounds leads to neutral mixed-metal trimeric building blocks that are used to produce the titled MOF via acetate/ligand exchange. | ||
An alternative method for the synthesis of mixed-valence oxo-trimer based MOFs consists in substituting, in a controlled manner, some of the constitutive iron(III) cations by other octahedrally coordinated metal cations to have a direct access to two valence states i.e., without requiring harsh activation conditions. Strategies to obtain mixed metal cation based solids have already been applied in MOF chemistry but in most cases consisted in (i) associating different cations of the same charge within the same building unit or (ii) cations of different charges but with different coordination numbers.14 Post-synthetic modification, such as metalation, is another attractive way to build mixed-metal materials, but this method suffers from a lack of control of the stoichiometry between the two different cations.15 However, if one attempts to prepare a mixed valence MOF from a direct mixture of iron(III) and metal(II) precursors, one would face the difficulty to prepare materials with the right architecture and a controlled iron(III)/metal(II) ratio. Besides, an elegant method to reach a higher degree of control of the synthesis consists in the use of pre-defined secondary building units (SBUs) to design porous solids with a controlled architecture. This method was previously reported for the design of porous iron(III) dicarboxylate MOFs (MIL-88s)7,16 or zirconium(IV) dicarboxylate solids (UiO-66's).17 It was proven that the structural integrity of the molecular building blocks was retained throughout the synthesis.18 Here, using oxo-centered trimeric mixed iron(III)/metal(II) acetates (M = Co, Ni, Mg) as building blocks, we report the scalable and green preparation of mixed-metal MOFs and show how it strongly impacts the accessibility of their Lewis acid sites. To illustrate our concept, we first applied this method to the rigid microporous iron(III) azobenzenetetracorboxylate MIL-127 or soc-MOF19,4a (Fig. 1). This solid with general formula Fe3O(C12N2H6(CO2)4)3/2X(H2O)2·n(solvent) (X = OH or Cl) combines several interesting aspects suited for practical applications. The framework is hydrothermally stable with iron(III) metal sites which are accessible upon departure of the coordinated water molecules. This MOF can be prepared easily and scaled-up under reflux in an environmentally friendly solvent, isopropanol.20 The preparations of nano to microscale MOFs are described and the performances of the materials prepared from different synthetic routes are compared by the authors.20 The material prepared with an environmentally friendly solvent possesses a rather high surface area (SBET > 1300 m2 g−1), well within the range of the surface areas obtained using different synthesis conditions. MIL-127 is therefore able to host biologically relevant cargoes21,22 and its iron(III) metal sites are beneficial for applications such as biomedicine,22 separation3b or catalysis.23
A series of mixed-metal MIL-127 materials with tailored properties is prepared. We primarily synthesized mixed-metal acetate building blocks using conditions adapted from previously reported protocols24 (Fig. 1 and Fig. S1, ESI†) i.e., neutral mixed metal building blocks with general formula FeIII2MIIO(H2O)2[O2C–CH3]6·nH2O (with M = Co or Ni). Furthermore, a method for the preparation of Fe/Mg acetate trimeric building blocks was developed (see ESI† for synthesis protocol details). The mixed metal acetate building blocks have then been used as precursors for the synthesis of the Fe/Co, Fe/Ni and Fe/Mg MIL-127 MOFs (Fig. 1 and ESI†) in environmentally friendly solvents (water, isopropanol) under reflux conditions. Such conditions allowed the easy upscaling of the preparation, producing materials with a space-time-yield of about 45 Kg m−3 d−1 (about 200 g could easily be obtained using a 5 L reactor). The formation of MIL-127 solids is confirmed by X-ray powder diffraction (XRPD) (see Fig. S2–S5, ESI†); Energy Dispersive X-ray spectroscopy (EDX) semi-quantitative analysis (see ESI†) indicated that, in all cases, the controlled iron to metal(II) stoichiometry of ca. 2 to 1 was obtained. Thermal gravimetric analysis (TGA), infrared (IR) spectroscopy and nitrogen sorption analysis were carried out to confirm the purity and integrity of the titled solids (Fig. S8–S10, ESI†), all of them presenting similar surface areas (∼1400 m2 g−1) well within the expected range. It is worth mentioning that direct synthetic approaches using a 2 to 1 ratio of iron(III) and metal(II) salts (chlorides), using similar synthesis conditions, led to either amorphous or recrystallized linkers. This confirms the fact that metal(III) and metal(II) cations do not possess the same chemical reactivity, as expected from the differences in terms of their acidity and metal–ligand bond lability.
57Fe Mössbauer spectroscopy data were collected at 300 K and 77 K on the mixed-metal MIL-127 MOFs as well as the corresponding starting mixed-metal acetate building block (see the spectra at 300 K Fig. S13, ESI†). The quadrupolar spectra of both series, which consist of broadened and asymmetrical lines, can be described by means of at least 2 components attributed to HS Fe3+ species: the comparison of their hyperfine structure reveals some similarities with, however, clearly larger quadrupolar splitting values in the case of mixed-metal MIL-127 MOFs. This is due to the presence of the ligand, which distorts the symmetry around the Fe probe. Nevertheless, it can be concluded from those hyperfine structures that the starting mixed-metal building blocks remain stable during the synthesis. To further confirm that our method allows the preparation of mixed-metal cation MOFs at the microscopic level, in situ infrared spectroscopy analysis was carried out on MIL-127(Fe, Ni) (Fig. 2). NO adsorption experiments, monitored by in situ IR spectroscopy, have been performed since it has been well established that the ν(NO) bands of adsorbed nitrosyls depend on the nature and the oxidation state of the accessible cations. Room temperature NO adsorption on the MIL-127(Fe, Ni) sample activated at 423 K (Fig. 2a) revealed two ν(NO) bands centered at about 1874 and 1797 cm−1. The band at 1797 cm−1 indicates the presence of NO adsorbed on Fe2+ sites.22 After activation at 503 K, the intensity of this band slightly increased (Fig. 2a′). It is noteworthy that the sharp ν(NO) band at 1874 cm−1 has not been observed for pure iron based MOFs. Its wavenumber is close to that reported for NO interaction with the Ni2+ cations supported on metal oxides25 and therefore, it is assigned to NO interacting with coordinatively unsaturated Ni2+ sites. When NO was adsorbed at low temperature (Fig. 2(b) and (b′)), an additional sharp band, observed at 1895 cm−1, can be assigned to the presence of NO weakly interacting with Fe3+.22 The concentration of NO adsorbed on the cation(+II) species was estimated from their molar absorption coefficient (see Fig. S11 and S12, ESI†). The number of Fe2+ sites is equal to about 70 μmol g−1 on MIL-127(Fe, Ni) activated to 423 K, and 210 μmol g−1 on MIL-127(Fe, Ni) activated to 503 K. However, the band intensity of NO/Fe2+ remained much lower on MIL-127(Fe, Ni) than on MIL-127(Fe)22 whatever the activation temperature (Fig. 2(a) and (a′)) showing that the fraction of reduced Fe sites is low in the mixed metal MOFs (less than 0.08 after activation at 423 K). By contrast, the number of NO/Ni2+ species (band at 1874 cm−1) was much more important. This amount is equal to about 600–800 μmol g−1 for these activation temperatures which implies that most cation(+II) sites were occupied by nickel. The global amount of (Fe, Ni)2+ was nearly constant after activation at 423 or 503 K. Considering that the reducible character of the iron(III) metal sites in MIL-100(Fe) or MIL-127(Fe) was related to the departure of the anion (F− or OH−) compensating the positive charge of the framework, the low reducible character of MIL-127(Fe, Ni) is in agreement with the expected neutral framework in mixed metal frameworks due to the presence of a divalent compensating cation in the trimer units. One could therefore attribute here the presence of a weak residual amount of Fe(II) centers for the mixed cation solids to defects or surface sites.
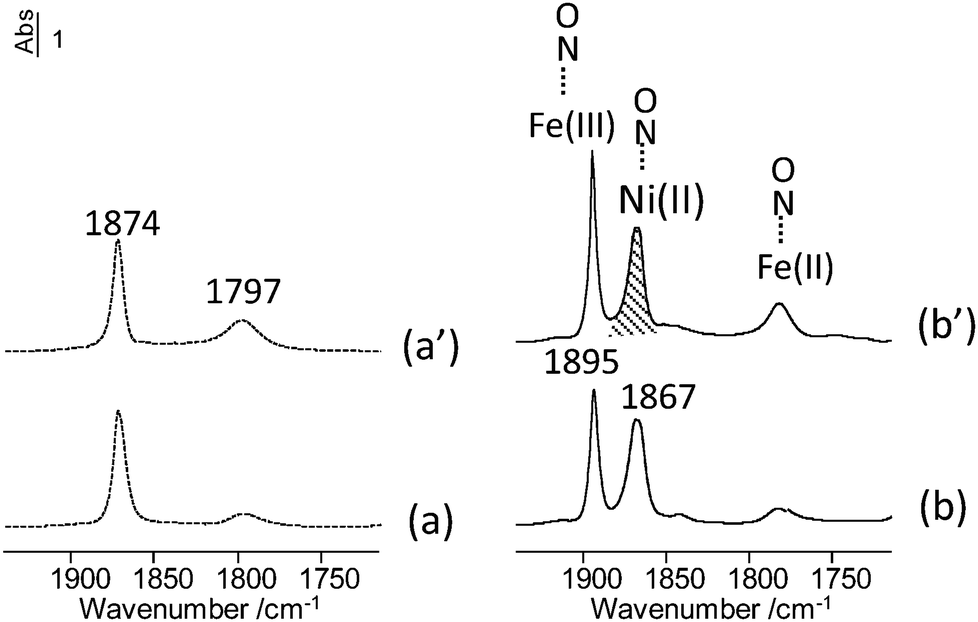 | ||
| Fig. 2 IR spectra of MIL-127(Fe, Ni) recorded after introduction of an equilibrium pressure of NO (665 Pa) at room temperature (dotted line) or low temperature (150 K, full line). Spectra (a) and (b): samples activated under secondary vacuum for 3 hours at 423 K. Spectra (a′) and (b′), samples activated under secondary vacuum during 6 hours at 503 K. | ||
Finally, to demonstrate the impact of the presence of the metal(II) cation within the porous architecture of the metal carboxylates, a combinatorial gas sorption analysis as well as adsorption microcalorimetry (see ESI†) using CO2 and CO gas probes26 were performed for the series of metal(II) substituted MIL-127 solids, which are also compared to the pure MIL-127(Fe). Fig. 3 shows the CO adsorption isotherms for MIL-127(Fe) and MIL-127(Fe, Ni) solids after activation to 423 K for 16 hours under vacuum. Indeed, previous studies have confirmed the presence of only Fe(III) metal sites upon activation at this temperature.22,30 It is clear that a rather low CO sorption capacity is observed in this pressure range for the pure iron solid, whereas the mixed Fe/Ni MIL-127 solid activated under the same conditions possesses a six-fold larger sorption capacity.
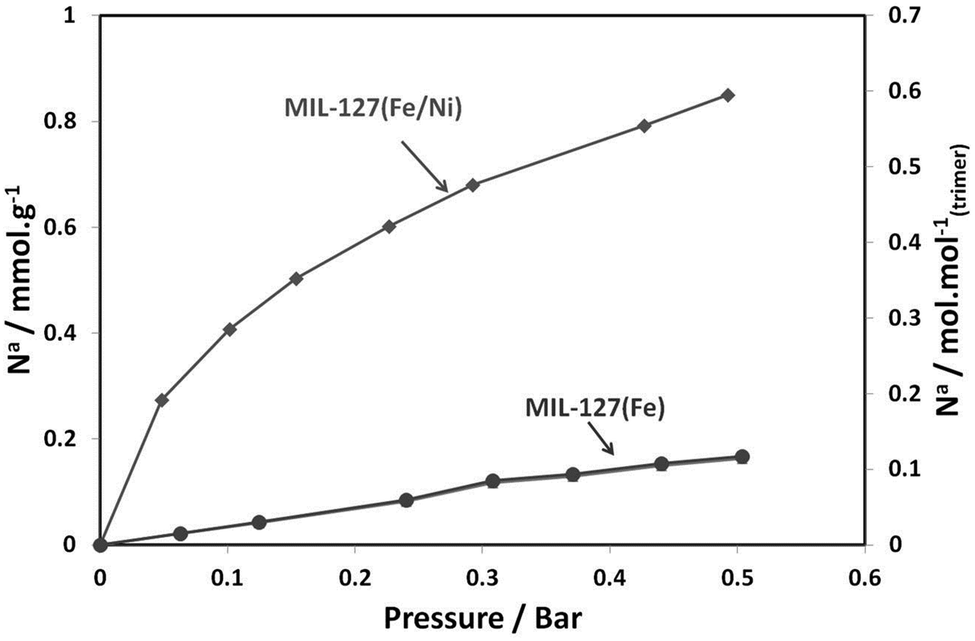 | ||
| Fig. 3 Adsorption isotherms for carbon monoxide in MIL-127(Fe, Ni) and MIL-127(Fe) at 303 K. Solids were activated at 423 K for 16 h under vacuum. | ||
Upon activation at higher temperature (473 K), a clear dependence of the nature of the metal(II) cation is noticeable for the polar CO probe (Fig. S15, ESI†). Activation to 473 K can lead to the formation of a significant amount of iron(II) sites.22,27,28 One can therefore deduce an affinity ranking for CO for the various divalent species with the trend: Ni2+ > Co2+ > Fe2+ > Mg2+, in agreement with the differences in the enthalpies of adsorption that lie from about −30 to −50 kJ mol−1 at low coverage (Fig. S15a, ESI†). This order of affinity for the metal(II) sites and CO is in agreement with previous data reported for CPO-27 or MOF-74.29 Indeed, this can be rationally explained by the electronegativity of these metals, following what is commonly observed through the back donation effect between metal(II) sites and unsaturated molecules. Whilst increasing the activation temperature of the MIL-127(Fe) solid leads to an increase in the CO adsorption capacity (see Fig. 3 and Fig. S15b, ESI†), this is not the case for the mixed valence materials. This trend is consistent with the IR data, which show for MIL-127(Fe) an increase of Fe2+ only with increasing activation temperature whilst for MIL-127(Fe, Ni) the amount of (Fe, Ni)2+ is not sensitive to the activation temperature. This suggests an easier availability of the unsaturated metal(II) sites of the mixed metal MIL-127 solids, since their accessibility only requires the departure of the coordinated solvent molecules, which generally occurs at lower temperature than the higher activation temperature necessary to create unsaturated metal(II) sites in the case of the pure iron MIL-127 (473 K). This represents an added value for practical applications, which often prefer low energetic activation conditions. Nonetheless, no significant differences are observed at low coverage (<1 bar) for CO2 (Fig. S14, ESI†). Finally, this strategy can be extended to other trimeric building unit based MOFs such as the highly flexible metal terephthalate MIL-88B7 or the rigid mesoporous trimesate MIL-10031 based on the Fe/Ni building block, which were successfully synthesized using our strategy (see synthesis and characterization details in the ESI†) as well as other mixed metal MOFs reported during the course of our study.32
Here, we firstly report the environmentally-friendly and scalable preparation of mixed iron(III)/metal(II) (M = Ni, Co, Mg) acetate building units for the design of mixed metal Fe(III)/M(II) (M = Co, Ni, Mg) porous MOFs of interest with a controlled iron/metal stoichiometry. IR spectroscopy and adsorption studies both confirm a direct accessibility of both Fe(III) and M(II) Lewis acid sites. Further studies will focus on the establishment of a library of mixed-metal MOFs and the analysis of their properties for various relevant applications.
The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 228862. The authors would also like to thank Antoine Tissot and Nathalie Guillou for their valuable help.
Notes and references
- Themed issues: Chem. Soc. Rev., 2014, 43, 5403–6176 Search PubMed; Chem. Rev., 2012, 112, 673–1268 Search PubMed; CrystEngComm, 2015, 17, 695 Search PubMed.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 15834 CrossRef CAS PubMed.
- (a) Y.-K. Seo, J. W. Yoon, J. S. Lee, Y. K. Hwang, C.-H. Jun, J.-S. Chang, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, S. Bourrelly, P. L. Llewellyn, P. Horcajada, C. Serre and G. Férey, Adv. Mater., 2012, 24, 806 CrossRef CAS PubMed; (b) J. Yoon, Y. K. Seo, Y. Hwang, J. S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. Llewellyn, C. Serre, P. Horcajada, J. M. Grenèche, A. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949 CrossRef CAS PubMed.
- (a) A. Dhakshinamoorthy, M. Alvaro, H. Chevreau, P. Horcajada, T. Devic, C. Serre and H. Garcia, Catal. Sci. Technol., 2012, 2, 324 RSC; (b) A. Dhakshinamoorthy, M. Alvaro, Y. K. Hwang, Y.-K. Seo, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 10719 RSC.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS PubMed.
- T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096 CrossRef CAS PubMed.
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284 RSC.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820 RSC.
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568 CrossRef CAS PubMed.
- D. Yuan, R. B. Getman, Z. Wei, R. Q. Snurr and H.-C. Zhou, Chem. Commun., 2012, 48, 3297–3299 RSC.
- S. B. Choi, M. J. Seo, M. Cho, Y. Kim, M. K. Jin, D.-Y. Jung, J.-S. Choi, W.-S. Ahn, J. L. C. Rowsell and J. Kim, Cryst. Growth Des., 2007, 7, 2290–2293 CAS.
- A. Fateeva, S. Devautour-Vinot, N. Heymans, T. Devic, J.-M. Grenèche, S. Wuttke, S. Miller, A. Lago, C. Serre, G. De Weireld, G. Maurin, A. Vimont and G. Férey, Chem. Mater., 2011, 23, 4641 CrossRef CAS.
- K. Wang, D. Feng, T.-F. Liu, J. Su, S. Yuan, Y.-P. Chen, M. Bosch, X. Zou and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983 CrossRef CAS PubMed.
- (a) F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J.-M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237 RSC; (b) A. D. Burrows, CrystEngComm, 2011, 13, 3623 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933 RSC.
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef PubMed.
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Férey, Chem. Commun., 2010, 46, 767 RSC.
- S. Surblé, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518 RSC.
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278 CrossRef CAS PubMed.
- H. Chevreau, et al. , J. Mater. Chem. Search PubMed , submitted.
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaïme, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767 CrossRef CAS.
- J. F. Eubank, P. S. Wheatley, G. Lebars, A. C. Mckinlay, H. Leclerc, P. Horcajada, M. Daturi, A. Vimont, R. E. Morris and C. Serre, APL Mater., 2014, 2, 124112 CrossRef PubMed.
- C. Serre, P. Horcajada, G. Férey, A. Vimont, M. Daturi, P.L. Llewellyn, Y.K. Hwang, J.-W. Yoon, J. S. Chang, 106660/FR PCT/FR2010/000402 (28/05/2010).
- H. Vrubel, T. Hasegawa, E. de Oliveira and F. S. Nunes, Inorg. Chem. Commun., 2006, 9, 208 CrossRef CAS PubMed.
- K. I. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42(1&2), 71 CAS.
- (a) H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2013, 343(6167), 167 CrossRef PubMed; (b) E. D. Bloch, M. R. Hudson, J. A. Mason, S. Chavan, V. Crocella, J. D. Howe, K. Lee, A. L. Dzubak, W. L. Queen, J. M. Zadrozny, S. J. Geier, L.-C. Lin, L. Gagliardi, B. Smit, J. B. Neaton, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 10752 CrossRef CAS PubMed.
- C. Serre, P. Horcajada, G. Férey, A. Vimont, M. Daturi, Y. K. Hwang, J. W. Yoon, J. S. Chang use of a porous crystalline hybrid solid as a nitrogen oxide reduction catalyst and devices, PCT/FR2010/000402 filed 28/05/2010.
- M. Maes, M. Trekels, M. Boulhout, S. Schouteden, F. Vermoortele, L. Alaerts, D. Heurtaux, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, I. Beuroies, R. Denoyel, K. Temst, A. Vantomme, P. Horcajada, C. Serre and D. E. De Vos, Angew. Chem., Int. Ed., 2011, 50, 4210 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, K. Sillar and J. Sauer, J. Phys. Chem. C, 2011, 115, 21777 CAS.
- S. Wuttke, P. Bazin, A. Vimont, C. Serre, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, G. Férey and M. Daturi, Chem. – Eur. J., 2012, 18, 11959 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef PubMed.
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and activation; X-ray powder diffraction; TGA; infrared, energy-dispersive X-ray and Mössbauer spectroscopies; N2, CO2 and CO sorption isotherms. See DOI: 10.1039/c5cc02550h |
| This journal is © The Royal Society of Chemistry 2015 |