
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the tunable surface chemistry of graphene oxide†
Zhen
Liu
a,
Jingquan
Liu
b,
Da
Li
b,
Paul S.
Francis
a,
Neil W.
Barnett
a,
Colin J.
Barrow
a and
Wenrong
Yang
*a
aCentre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Deakin University, Geelong, VIC 3216, Australia. E-mail: wenrong.yang@deakin.edu.au
bCollege of Chemical Science and Engineering, Laboratory of Fiber Materials and Modern Textile, The Growing Base for State Key Laboratory, Qingdao University, Qingdao, China
First published on 2nd June 2015
Abstract
The determination of oxygen content, hydrophobicity and reduction efficiency of graphene oxide (GO) are difficult tasks because of its heterogeneous structure. Herein, we describe a novel approach for the detailed understanding of the surface chemistry of GO by studying the interactions between [Ru(bpy)3]2+ and GO.
Despite its short history since 2004, graphene has been widely explored through functionalising this extraordinary two-dimensional material.1,2 Among various derivatives, graphene oxide (GO) is the most popular form of functionalised graphene, which incorporates carboxylic, hydroxyl and carbonyl groups at its edges, and epoxy and hydroxyl groups on its basal plane.3,4 These oxygen-containing groups impart a negative surface charge that inhibits irreversible sheet aggregation in solution.5 The corresponding processibility and monolayer dispersion in aqueous solution are the most attractive points to researchers, as well as the further chemical modification sites they provide, allowing, for example, amidation reactions.6 Although GO has gained considerable application value in many areas,7,8 more work is still being performed to improve its surface properties via chemical reductions,9 particularly due to conductivity problems.10,11 There are many reducing agents that could functionalise graphene according to well-supported mechanisms or proposed mechanisms based on knowledge of organic chemistry.12 Determination of the amount of different oxygen-containing groups and the hydrophobicity of GO surface, and evaluation of the efficiency of chemical reactions are still difficult tasks because of the heterogeneous structure of GO.13L-Ascorbic acid, which possesses a mild reducing capacity and nontoxic nature, is frequently applied as a reductant in biochemistry.14 Herein we present a detailed surface chemistry study of graphene oxide using tris(2,2′-bipyridine)ruthenium(2+) dichloride ([Ru(bpy)3]Cl2) as a detection probe, exploring the interactions between GO and this complex. [Ru(bpy)3]2+ has been reported to be able to functionalise various matrixes via electrostatic absorption15 and hydrophobic interactions.16 A controlled chemical reduction of GO by L-ascorbic acid was carried out to adjust the oxygen content on its surface. This was monitored by the transition of surface interactions of the chemically reduced graphene oxide (CRGO).
Our hypothesis for this work is displayed in Fig. 1. The GO surface is full of oxygen functional groups, but there is a gradual removal of oxygen by the controlled chemical reduction. Not surprisingly, the surface charge (−) of GO will drop, and therefore the hydrophobicity of the carbon nanosheets will be restored to some extent.17 This control of surface properties facilitates convenient binding of graphene materials with various modifiers via covalent or non-covalent binding.18
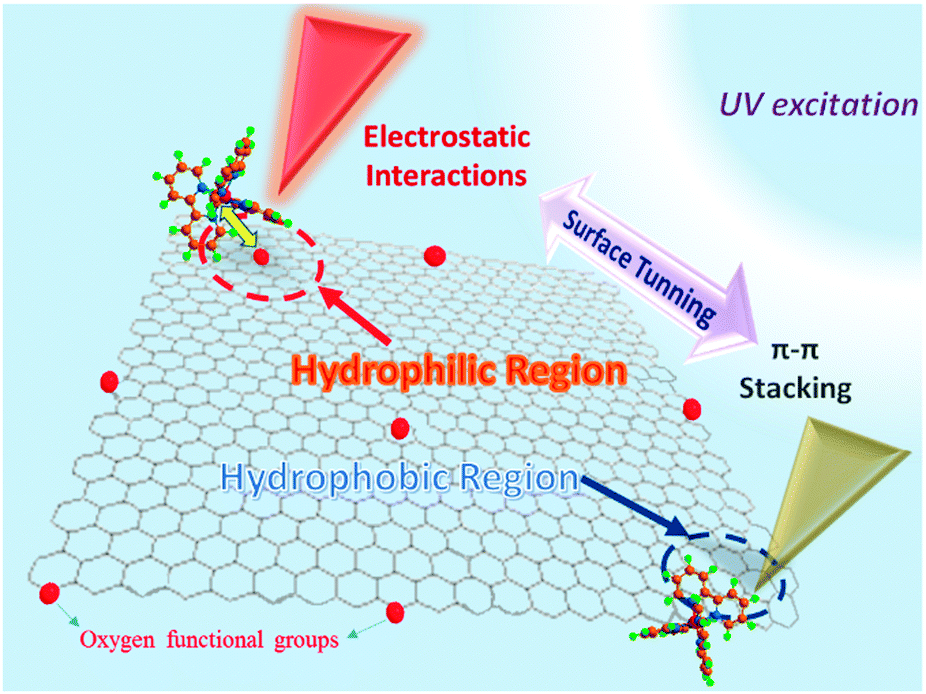 | ||
| Fig. 1 Schematic of the surface chemistry tuning of graphene oxide. [Ru(bpy)3]2+ was used as the probe to observe the change in properties on the graphene surface, by monitoring the transformation of dominate interactions in CRGO–[Ru(bpy)3]2+ composites. | ||
[Ru(bpy)3]2+ is amphipathic molecule with three bipyridine ligands binding to a ruthenium metal center,19 in a three-dimensional spatial arrangement (see ESI,† Fig. S1). We selected [Ru(bpy)3]2+ as the modification molecule because it is able to probe the graphene surface through two categories of non-covalent binding, namely electrostatic interaction and π–π stacking.20,21 Interestingly, the mechanisms of these two binding categories are somewhat contradictory, with the former based on surface charge, while the latter is dependent on the hydrophobicity of the graphene sheets. Thus, we examined the interactions that existed between GO and [Ru(bpy)3]2+ for a better understanding of its surface chemistry. Self-assembly was applied to prepare all of the samples by simple chemical mixing and slight shaking.
Firstly, UV-vis absorbance spectra (Fig. 2a) showed that the absorbance intensities of [Ru(bpy)3]2+–GO hybrids were positively correlative to the dosage of GO. However, the peak at 456 nm, attributed to the metal-to-ligand transition of [Ru(bpy)3]2+, was decreased by GO binding. This is due to the new pathway of electron transfer introduced by GO, in which the Ru metal center directly delivered an electron to the GO sheets via COOH or OH, indicative of their electrostatic interaction.22 This was further characterized by a zeta potential test (see ESI,† Fig. S2). The addition of [Ru(bpy)3]2+ into GO solutions caused the sudden decline of surface charge ((−) from GO). This simultaneously brought a sharp increase in particle size (almost double) due to sheet aggregation.
 | ||
| Fig. 2 Characterizations of GO–[Ru(bpy)3]2+ composites to study the internal interactions. UV-vis absorbance spectra (a) and phosphorescence emission spectra (b) (excited at 286 nm) of GO–[Ru(bpy)3]2+ mixture with the same dosage of [Ru(bpy)3]2+ but increasing GO concentrations (shown by black arrow). All of the experiments were carried out in a basic solution (pH 9.5); with supporting ATR-FTIR (c) and XRD (d) results of GO–[Ru(bpy)3]2+ composite materials which were made into thin films by vacuum filtration. | ||
On the other hand, photoluminescence quenching provided strong evidence of π–π stacking interactions on the graphene surface.23 The orange phosphorescence (λmax ∼ 610 nm) from ruthenium(II) diimine complexes arises from the excitation of an electron from the metal-based d(πM) orbitals to ligand-based π* antibonding orbitals,24 (i.e. metal-to-ligand charge-transfer, MLCT), followed by intersystem crossing to the lowest excited triplet state.25 The adhesion of the bipyridine ligands on GO sheets via π–π stacking introduces a new pathway of electron-transfer from the Ru metal center to GO π-orbitals, inhibiting the generation of phosphorescence. Although GO (prior to chemical reduction) contained many oxygen functional groups, it was still able to quench the phosphorescence of [Ru(bpy)3]2+ due to π–π stacking on its hydrophobic regions (Fig. 2b). With an increase of GO dosage, there will be less and less free [Ru(bpy)3]2+ in the mixture, leading to the stronger quenching of phosphorescence. Therefore, strong π–π stacking was observed between the GO surface and the bipyridine ligands of [Ru(bpy)3]2+. In Raman spectra (see ESI,† Fig. S3), we found a slight decrease of ID/IG due to the fixing of [Ru(bpy)3]2+ on the GO surface, as the π–π stacking extended the π-conjugated structure corresponding to G band intensity.26
Attenuated total reflectance – Fourier transform infrared spectroscopy (ATR-FTIR) verified the successful self-assembly of [Ru(bpy)3]2+ on GO (Fig. 2c). The pattern of GO (black curve) showed characteristic peaks at 1722 cm−1 and 1614 cm−1, respectively, both of which experienced a blue-shift after the composite material was formed (blue curve). This indicated the addition of [Ru(bpy)3]2+ affected the chemical environment of both COOH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, with the Ru metal center close to COO− groups and bipyridine ligands attached on the CC conjugated structure. The GO and the composite were made into films for X-ray diffraction (XRD) analysis (Fig. 2d). The GO exhibited a sharp (002) peak at 10.6° revealing a d-spacing of 0.83 nm between the adjacent sheets. When introducing the ruthenium complex, this peak shifted to 9.3° and became weaker, which was attributed to increased interlayer spacing and disorder. Through these results, the interactions between GO and [Ru(bpy)3]2+ have been shown to include both electrostatic and π–π stacking interactions, and electron transfers between them have also been studied.
C bonds, with the Ru metal center close to COO− groups and bipyridine ligands attached on the CC conjugated structure. The GO and the composite were made into films for X-ray diffraction (XRD) analysis (Fig. 2d). The GO exhibited a sharp (002) peak at 10.6° revealing a d-spacing of 0.83 nm between the adjacent sheets. When introducing the ruthenium complex, this peak shifted to 9.3° and became weaker, which was attributed to increased interlayer spacing and disorder. Through these results, the interactions between GO and [Ru(bpy)3]2+ have been shown to include both electrostatic and π–π stacking interactions, and electron transfers between them have also been studied.
The chemical reductions of GO were carried out using L-ascorbic acid as a reducing agent, and were recorded in real-time by UV-vis and Raman spectra (see ESI,† Fig. S4). Five different CRGO samples were synthesized by controlling the reduction time (1 h, 6 h, 12 h, 24 h and 48 h). Stepwise red-shifts of the GO peak (230 nm) were seen in the UV-vis spectra (Fig. S4a, ESI†), corresponding to gradual removal of oxygen containing groups (also evidenced by the thickness decrease in atomic force microscope (AFM) results (Fig. S5a, ESI†)). However, even the most reduced sample (48 h, peak centered at 244 nm) retained oxygen groups when compared with highly reduced GO, which commonly exhibits maximum absorbance at 268 nm.27 In Raman spectra (Fig. S4b, ESI†), there was an obvious increase of D band intensity, due to the introduction of more defects in the graphene sheets during reductions. We would suggest that the as-prepared CRGOs should have similar functions as GO in terms of binding methods; however, we needed to ascertain what happened on their surface and how the properties changed.
First, in the UV-vis absorbance spectra (Fig. 3a), we observed a blue-shift in the 286 nm peak in [Ru(bpy)3]2+–CRGO composites, indicating an important change in the CRGO surface. This is strong evidence of the restoration of graphene's hydrophobicity, which means larger surface area, lower oxygen content, and stronger ability to bind by π–π stacking. Interestingly, this is achieved through spectral shift of probe molecule rather than direct detection of the graphene surface. The 286 nm peak of [Ru(bpy)3]2+ corresponded to the π to π* transition of the bipyridine ligands. This shift therefore demonstrated another type of π electron delocalization from π–π stacking with the GO surface. Importantly, we found that CRGOs subjected to longer reduction-time shifted this peak further (reaching 280 nm by the 48 h reduction sample), indicative of stronger π–π stacking. Obviously the restoration of hydrophobicity of the graphene material facilitated π–π stacking interactions on its surface. This was further verified using phosphorescence emission spectra (Fig. 3b), in which CRGOs showed different degrees of phosphorescence quenching. With the same [Ru(bpy)3]2+ concentration in all solutions, the more reduced sample proved to be the stronger quencher (from red curve down to the dark-blue curve and from left to right in the inset), again corresponding to greater π–π stacking.
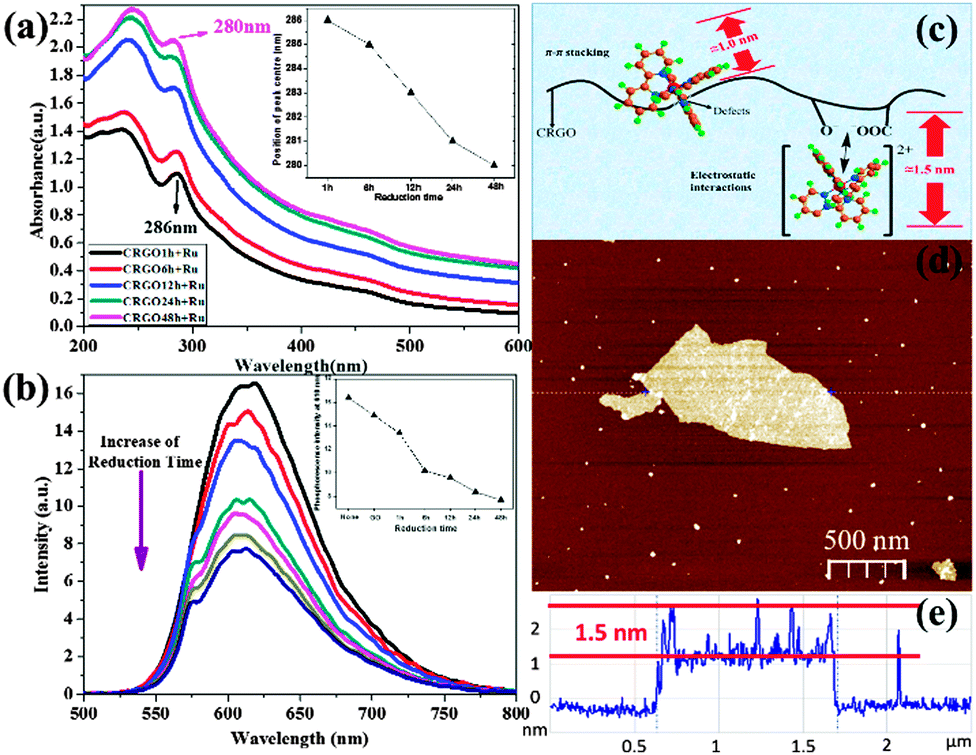 | ||
| Fig. 3 Characterizations of graphene surface tuning using [Ru(bpy)3]2+ as a probe. UV-vis absorbance (a) and phosphorescence emission (b) (excited at 286 nm) spectra of [Ru(bpy)3]2+ bound with CRGO under different reduction times (1 h, 6 h, 12 h, 24 h and 48 h). All of the experiments were carried out in a basic solution (pH 9.5); Schematic figure (c) to show our speculation about the thickness of the [Ru(bpy)3]2+ monolayer on graphene via electrostatic interactions and π–π stacking, and AFM image (d) and corresponding height profile (e) of [Ru(bpy)3]2+ on the GO sheet. | ||
In Fig. 3c, we present our viewpoint of the morphology of the [Ru(bpy)3]2+ monolayer bound to GO, referring to our AFM results. The thickness of [Ru(bpy)3]2+ on the GO sheets ranged from 1.0 nm to 1.5 nm (Fig. 3d and e). We suggest height fluctuations might be caused by the influence of different modes of binding. [Ru(bpy)3]2+ has a 3D structure (shown in Fig. S1b, ESI†) and thus a spatial barrier while assembling. When it is attached to hydrophobic regions of graphene via π–π stacking interactions, it could be expected to remain at the edges and near defects, where the spatial barrier was minimized. Significantly, one of its ligands must sit parallel with the graphene sheet in order for the π electron hybridization.
We suppose that this type of binding might make its thickness (around 1.0 nm) lower than those linked via electrostatic interactions (around 1.5 nm). The latter took place on oxygen functional groups that had already imparted an increase in height onto the graphene sheets. To examine this notion, monolayer CRGO sheets were obtained on mica substrates by spin coating. In order to get a single layer of [Ru(bpy)3]2+ on the CRGO surface, a low concentration [Ru(bpy)3]2+ solution (10 μM) was dropped on the CRGO coated mica surface, kept for 60 s, and then absorbed by wiping paper. The thicknesses of the [Ru(bpy)3]2+ molecules on different CRGO samples were determined using AFM (see ESI,† Fig. S5b) and corresponding height profiles. The results for the height range and average heights are presented in Table 1. In this work, we wanted to focus on monolayer [Ru(bpy)3]2+ on graphene sheets, thus we ignored those areas where a few layers of [Ru(bpy)3]2+ flocked together. The measured sections exhibited a height range from 1.0 nm to 1.5 nm, which verified both electrostatic and π–π interactions, consistent with our deduction. Moreover, a decrease in average height from 1.4 nm to 1.2 nm was observed due to the transformation from electrostatic interactions to π–π stacking interactions by gradual reduction.
| Samples | Height range (nm) | Average height (nm) |
|---|---|---|
| CRGO 1 h + [Ru(bpy)3]2+ | 1.0–1.5 | 1.4 |
| CRGO 6 h + [Ru(bpy)3]2+ | 1.0–1.5 | 1.3 |
| CRGO 12 h + [Ru(bpy)3]2+ | 1.0–1.5 | 1.3 |
| CRGO 24 h + [Ru(bpy)3]2+ | 1.0–1.5 | 1.3 |
| CRGO 48 h + [Ru(bpy)3]2+ | 1.0–1.5 | 1.2 |
A loading experiment of [Ru(bpy)3]2+ on GO gave further insight into GO reduction (see ESI,† Fig. S6). As both the electrostatic and π–π interactions contributed to the binding of [Ru(bpy)3]2+ on graphene, its loading capacity would change during the reduction process. Untreated GO exhibited the greatest ability to accommodate [Ru(bpy)3]2+ (0.26 mg mg−1), due to the large amount of oxygen groups on its surface. The sharp decrease exhibited by the 1 h reduction-time sample showed that a large decrease in surface charge could be achieved within a short period of time. With longer reduction time, the loading began to rise and reached 0.21 mg mg−1 for the 48 h CRGO sample, corresponding to the most extensive restoration of hydrophobicity.
Cyclic voltammetry curves (Fig. S7a, ESI†) were used to investigate the electrochemistry of the graphene–[Ru(bpy)3]2+ composite. Due to the oxidation of [Ru(bpy)3]2+, there is an peak around 1190 mV. The introduction of graphene brought a negative shift of this peak, and the more reduced CRGO had the stronger ability to shift it, moving to 1102 mV in 48 h CRGO. We suggest that the decrease of oxidation potential may be attributed to the decrease of oxidation energy due to the enhanced electron transfer. The rate of heterogeneous electron transfer (HET), namely the transfer of electrons from/to graphene sheets to/from compounds, is related not only to the properties of binding molecules but also to the amount of defects and functional groups presented on graphene surface.28 We found that the enhanced electron-transfer kinetics could facilitate the redox reactions of [Ru(bpy)3]2+ that usually required high overpotentials to achieve. This could significantly lower the energy consumption in the electrochemical process.29 Herein, the reduction of GO helped to save energy costs by improving the HET rate.
For a better understanding of the influence of oxygen functionalities, peak-to-peak separations (ΔE) were plotted against reduction time of GO (Fig. S7b, ESI†). The decreasing trend shown in the figures indicated improvement of the HET by removal of oxygen functional groups.30 Furthermore, the restoration of sp2 carbon–carbon improved the conductivity of the modified electrode surface, which was evidenced by the increasing oxidation currents with longer reduction time.31 Based on the reduction peaks, we observed that the redox reaction of [Ru(bpy)3]2+/3+ is not reversible, especially in those less reduced samples (GO, 1 h CRGO, 6 h CRGO and 12 h CRGO). As discussed above, for GO and less reduced CRGO, electrostatic interactions are dominated through the binding of the ruthenium complex, and their electron transfers rely on negatively charged groups (like COO− and O−). In these samples, the free electrons used for Ru3+ reduction suffer from electrostatic repulsions from the negative charge of graphene surface, preventing them from approaching Ru3+, which remains very close to those oxygen functional groups. Therefore, the reduction of Ru3+ was inhibited, leading to lower reduction currents (observed for GO, and CRGO reduced for 1 h, 6 h and 12 h). However, this inhibiting effect would not occur when π–π stacking plays the major role in the binding (such as 24 h CRGO and 48 h CRGO) because the electrons transfer through graphene's conjugated region rather than oxidized sites. This explains why the reduction currents of the last two samples are comparable with their oxidation currents.
This novel analytical approach to probe the surface chemistry of GO has provided new insight into the properties of this important material. Chemical reduction for GO surface tuning was successfully employed to manipulate the surface charge and hydrophobicity, which enabled us to control the electrostatic and π–π stacking interaction. Our work presents a novel method to explore surface chemistry and to control binding modes on modified carbon materials.
The authors would like to thank Deakin University for financial support.
Notes and references
- K. Novoselov, A. Geim, S. Morozov, D. Jiang, Y. Zhang, S. Dubonos, I. Grigorieva and A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2009, 39, 228 RSC.
- Y. Chen, W.-C. Lin, J. Liu and L. Dai, Nano Lett., 2014, 14, 1467 CrossRef CAS PubMed.
- C. Cheng and D. Li, Adv. Mater., 2013, 25, 13 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554 CAS.
- C. Wu, Y. Zhou, X. Miao and L. Ling, Analyst, 2011, 136, 2106 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291 RSC.
- W. Ai, Z. Du, Z. Fan, J. Jiang, Y. Wang, H. Zhang, L. Xie, W. Huang and T. Yu, Carbon, 2014, 76, 148 CrossRef CAS PubMed.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed.
- N. Zhu, S. Han, S. Gan, J. Ulstrup and Q. Chi, Adv. Funct. Mater., 2013, 23, 5297 CrossRef CAS PubMed.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112 RSC.
- Z. Guo, Y. Shen, M. Wang, F. Zhao and S. Dong, Anal. Chem., 2003, 76, 184 CrossRef.
- Z. Guo and S. Dong, Anal. Chem., 2004, 76, 2683 CrossRef CAS PubMed.
- L. Baptista-Pires, B. Pérez-López, C. C. Mayorga-Martinez, E. Morales-Narváez, N. Domingo, M. J. Esplandiu, F. Alzina, C. M. S. Torres and A. Merkoçi, Biosens. Bioelectron., 2014, 61, 655 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- A. W. Knight, TrAC, Trends Anal. Chem., 1999, 18, 47 CrossRef CAS.
- H. Li, J. Chen, S. Han, W. Niu, X. Liu and G. Xu, Talanta, 2009, 79, 165 CrossRef CAS PubMed.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490 CrossRef CAS PubMed.
- Y. Yuan, H. Li, S. Han, L. Hu, S. Parveen, H. Cai and G. Xu, Anal. Chim. Acta, 2012, 720, 38 CrossRef CAS PubMed.
- Z. Liu, J. Liu, L. Cui, R. Wang, X. Luo, C. J. Barrow and W. Yang, Carbon, 2013, 51, 148 CrossRef CAS PubMed.
- d. M. Maykel, M. Álvaro and H. García, Langmuir, 2012, 28, 2849 CrossRef PubMed.
- G. J. Barbante, P. S. Francis, C. F. Hogan, P. R. Kheradmand, D. J. Wilson and P. J. Barnard, Inorg. Chem., 2013, 52, 7448 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- F. Thema, M. Moloto, E. Dikio, N. Nyangiwe, L. Kotsedi, M. Maaza and M. Khenfouch, J. Chem., 2013, 2013, 1 CrossRef.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150 CrossRef CAS PubMed.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Chem. – Eur. J., 2011, 17, 10763 CrossRef CAS PubMed.
- S. M. Tan, A. Ambrosi, C. K. Chua and M. Pumera, J. Mater. Chem. A, 2014, 2, 10668 CAS.
- W. Deng, X. Ji, M. Gómez-Mingot, F. Lu, Q. Chen and C. E. Banks, Chem. Commun., 2012, 48, 2770 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc02518d |
| This journal is © The Royal Society of Chemistry 2015 |