
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A short pyrene-fused pyrazaacene with red to near-infrared photoluminescence†
Raúl
García
a,
Manuel
Melle-Franco
b and
Aurelio
Mateo-Alonso
*ac
aPOLYMAT, University of the Basque Country UPV/EHU, Avenida de Tolosa 72, E-20018 Donostia-San Sebastian, Spain. E-mail: amateo@polymat.eu
bCentro ALGORITMI, 4710-057 Braga, Portugal
cIkerbasque, Basque Foundation for Science, Bilbao, Spain
First published on 16th April 2015
Abstract
A pyrene-fused pyrazaacene with only four linearly-fused aromatic rings is reported, which remarkably shows emission at red to NIR wavelengths (λem = 687 nm). This exceeds the emission wavelengths observed for azaacenes with the same number of linearly-fused rings.
Near infrared (NIR) emissive organic materials are receiving growing interest because of their potential applications in light-emitting diodes,1 sensing2 and bioimaging,2,3 among others. Notably, current optical communications use NIR wavelengths for transmission and processing. Also, NIR light penetrates deeper into biological tissues and is less damaging than visible and UV light, and thus offers potential for non-invasive studies within the body. New advances in these technologies depend on the development of NIR organic-based materials and our abilities to model their emission characteristics.
Pyrene-fused pyrazaacenes4 (PPAs) have recently attracted much attention as molecular materials for organic electronic applications due to their enhanced stability. NIR-emitting PPA can be in principle obtained by increasing the number of longitudinal fused rings. This is well illustrated in the n-azaacene5 series, in which a progressive redshift of the emission bands is observed with an increasing number of fused rings. Nevertheless, the emission of PPA4,6 occurs at considerably lower wavelengths than those of n-azaacenes5 with an equal number of linearly-fused rings (Fig. 1). For example, while an n-diazatetracene derivative 1 shows emission at yellow wavelengths (λem = 584 nm),7 the pyrene-fused diazatetracene 2 with the same number of linearly-fused rings emits at blue wavelengths (λem = 461 nm).8 Therefore, to obtain NIR-emitting PPA, a large number of linearly-fused rings would be necessary, which would imply an additional synthetic effort.
 | ||
| Fig. 1 Structure of n-azatetraacene 1 and pyrene-fused azatetraacenes 2 and 3. | ||
Herein, we report PPA 3 with only four linearly-fused aromatic rings (Fig. 1), which remarkably shows emission at red to NIR wavelengths (λem = 687 nm), exceeding the emission wavelengths observed not only for PPA but also for n-azaacenes with the same number of linearly-fused rings. This behaviour is consistent with an emissive charge transfer state, as a result of the highly polarised electronic structure of its aromatic core, which is corroborated by comparative spectroscopic and electrochemical studies with a series of carefully selected reference compounds together with theoretical calculations.
The synthesis of PPA 3 and reference compounds 4–6 was achieved following the route depicted in Scheme 1. Pyrene diketone 7 was obtained in two steps following a procedure reported by Harris.9t-Butyl groups in the 2,7 positions were introduced in order to enhance the solubility of the resulting compounds. Cyclocondensation of diketone 7 with 2,3-diamino-5,6-dicyanopyrazine provided pyrene-fused tetracene 3 with two cyano groups and two fused pyrazines in good yields (66%). To study the effects of the number of fused pyrazine rings on the overall optoelectronic properties, reference compound 5 with just one pyrazine ring was synthesised in good yields (65%) by means of cyclocondensation of diketone 7 with diaminomaleonitrile. Reference compounds 4 and 6 with a truncated conjugation along the K region (positions 9,10 of the pyrene core) were synthesised in order to study the effects on the optoelectronic properties. To do so, pyrene diketone 7 was first transformed into diketal-protected 8 and then was subsequently oxidised to pyrene diketone 9, following a previously reported procedure.10 Compounds 4 and 6 were obtained by cyclocondensation of diketone 9 with 2,3-diamino-5,6-dicyanopyrazine and diaminomaleonitrile in good yields (52% and 43%, respectively). Compounds 3–6 are soluble in a wide variety of organic solvents, including CH2Cl2, CHCl3 and THF.
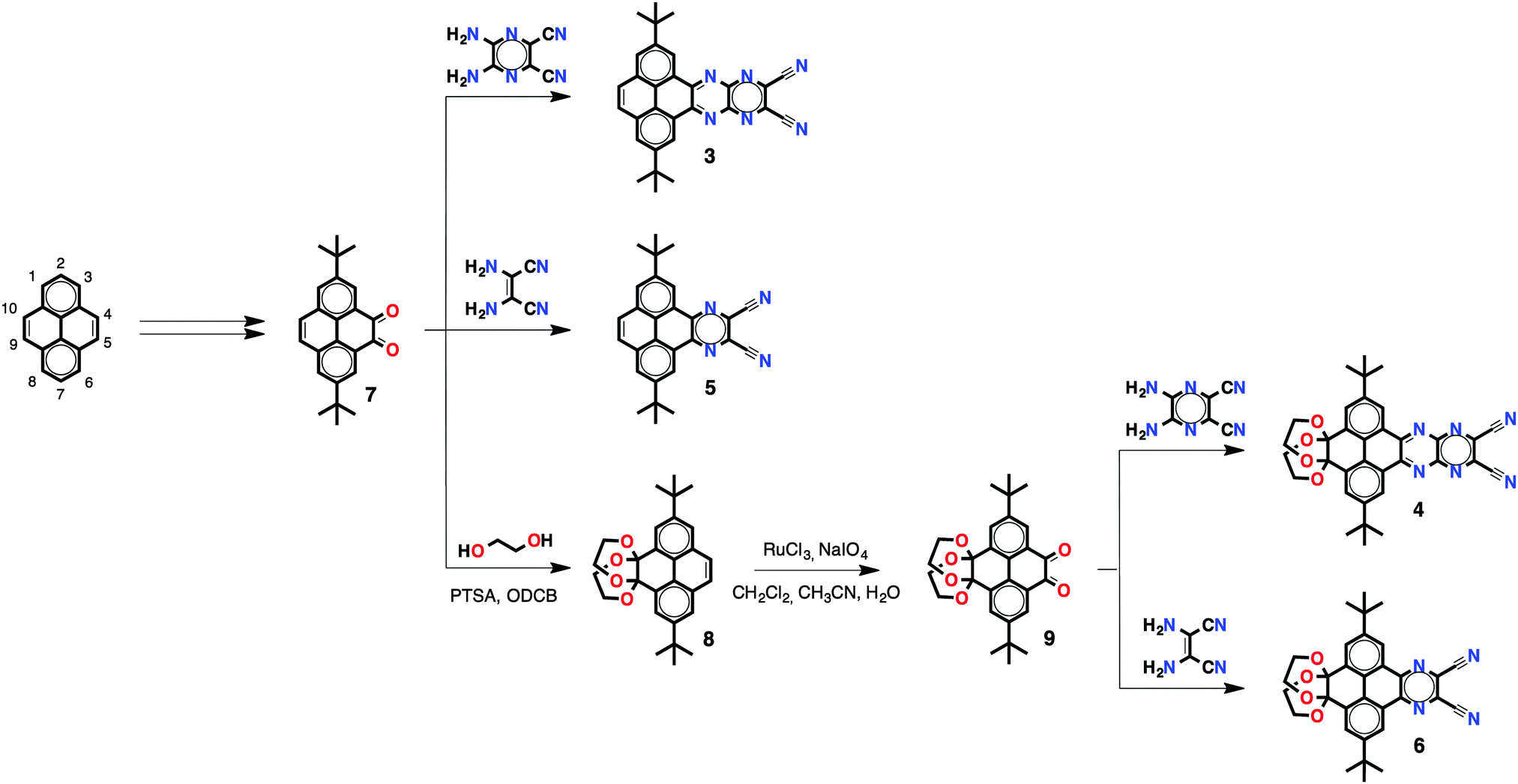 | ||
| Scheme 1 Synthesis of compounds 3–6. | ||
The absorption spectrum of 3 (Fig. 2) displays two narrow absorption bands centred at 354 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε = 4.31) and 366 nm (logε = 4.47) and a broad and intense absorption band centred at 571 nm (logε = 3.94). The latter is consistent with its intense purple coloration and also with an intramolecular charge transfer process as a result of the highly polarised electronic structure of its aromatic core. On the other hand, the K-truncated compound 4 exhibits a yellow coloration and absorption bands that are substantially blueshifted, as much as 126 nm, to 351 (logε = 4.25) and 445 nm (logε = 4.23). Compound 5 is orange and displays absorption bands centred at 364 (logε = 3.93) and 455 nm (logε = 3.44). The K-truncated compound 6 is pale yellow and its absorption spectrum shows similar bands that are blueshifted, as much as 65 nm, to 347 (logε = 4.45) and 390 nm (logε = 3.98).
ε = 4.31) and 366 nm (logε = 4.47) and a broad and intense absorption band centred at 571 nm (logε = 3.94). The latter is consistent with its intense purple coloration and also with an intramolecular charge transfer process as a result of the highly polarised electronic structure of its aromatic core. On the other hand, the K-truncated compound 4 exhibits a yellow coloration and absorption bands that are substantially blueshifted, as much as 126 nm, to 351 (logε = 4.25) and 445 nm (logε = 4.23). Compound 5 is orange and displays absorption bands centred at 364 (logε = 3.93) and 455 nm (logε = 3.44). The K-truncated compound 6 is pale yellow and its absorption spectrum shows similar bands that are blueshifted, as much as 65 nm, to 347 (logε = 4.45) and 390 nm (logε = 3.98).
 | ||
| Fig. 2 Normalised absorption spectra of 3–6 in CH2Cl2. | ||
The lower energy singlet excited states of 3–6 were calculated using time-dependent density functional theory (TD-DFT B3LYP-CH2Cl2-6311g+(d2,p)/B3LYP-CH2Cl2-631(d,p)) in order to shed light on the nature of the electronic transitions in the electronic spectra. The simulated spectra of the TD-B3LYP reproduce reasonably well the experimental spectra (see Fig. S1, ESI†), and most importantly, illustrate that truncating the conjugation along the K region deactivates significantly the HOMO → LUMO transition. TD-DFT predicts that the experimental band centred at 571 nm in 3 is described as an HOMO → LUMO transition (estimated at 633 nm) with a contribution from the HOMO−1 → LUMO transition (estimated at 587 nm), which shows a significantly reduced oscillator strength (∼22 fold) in comparison to the HOMO → LUMO transition. The picture is different for K-truncated 4, where the band centred at 445 nm corresponds to a HOMO−1 → LUMO transition (estimated at 463 nm). The HOMO → LUMO transition (estimated at 623 nm) shows a significantly reduced oscillator strength (∼41 fold) in comparison to the HOMO−1 → LUMO transition. Therefore the HOMO → LUMO transition is not noticeable in the experimental spectrum of 4. The experimental band centred at 455 nm in the spectrum of 5 is described as a HOMO → LUMO transition (estimated at 482 nm) with a HOMO → LUMO+1 contribution (estimated at 447 nm). The picture is again different for K-truncated 6. The experimental band centred at 390 nm corresponds to a HOMO−1 → LUMO transition (estimated at 377 nm). Remarkably, the HOMO → LUMO and the HOMO → LUMO+1 transitions (estimated, respectively, at 454 and 413 nm) show a significantly reduced oscillator strength (∼20 fold) in comparison to the HOMO−1 → LUMO transition. Therefore the HOMO → LUMO and HOMO → LUMO+1 transitions are not noticeable in the experimental spectrum of 6.
The photoluminescence spectra obtained for 3–6 (Fig. 3) show almost featureless emission bands with some vibronic shoulders. In all cases, the emission bands are almost mirror images of the corresponding absorption bands. Remarkably, while 4, 5, and 6 emit in the visible region, pyrene-fused tetracene 3 displays a featureless emission band centred at 687 nm that spans into the NIR region up to 860 nm. The emission bands of 5 are bathochromically shifted (24 nm) when compared with 6. The same trend was also observed in the emission spectra of 3 and 4 but at a much higher extent, as the emission bands of 3 appear to be bathochromically shifted (187 nm) when compared with those of 4. A progressively redshifted fluorescence is discernible when comparing 3 with 5 (151 nm) and 4 with 6 (14 nm), which is again consistent with the longitudinal extension of the π-system with an additional pyrazine ring.
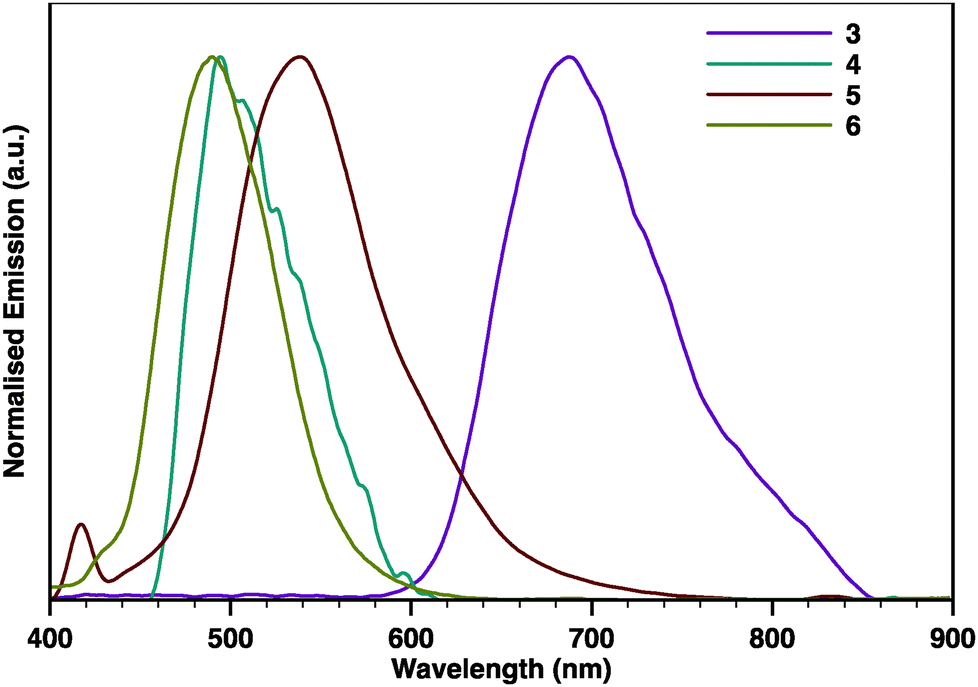 | ||
| Fig. 3 Normalised photoluminescence spectra of 3–6 in CH2Cl2. | ||
Cyclic voltammetry evidences the electron-deficient nature of 3–6 and also substantial differences in their redox properties (Fig. 4). The extended pyrene-fused tetracenes 3 and 4 present a complex redox behaviour. Two reduction waves are observed for 3. However, three reduction waves are observed for 4; two of them coincide with those observed for 3, while an additional reduction wave with double current intensity appears in between. PPAs 5 and 6 display one reversible reduction wave at ∼0.9 V that is also present for 3 and 4. According to the first half-wave potential (EI1/2), pyrene-fused tetracenes 3 and 4 are much easier to reduce than PPAs 5 and 6, which is in agreement with the extra pyrazine rings that stabilise considerably the electrochemically-generated anions.
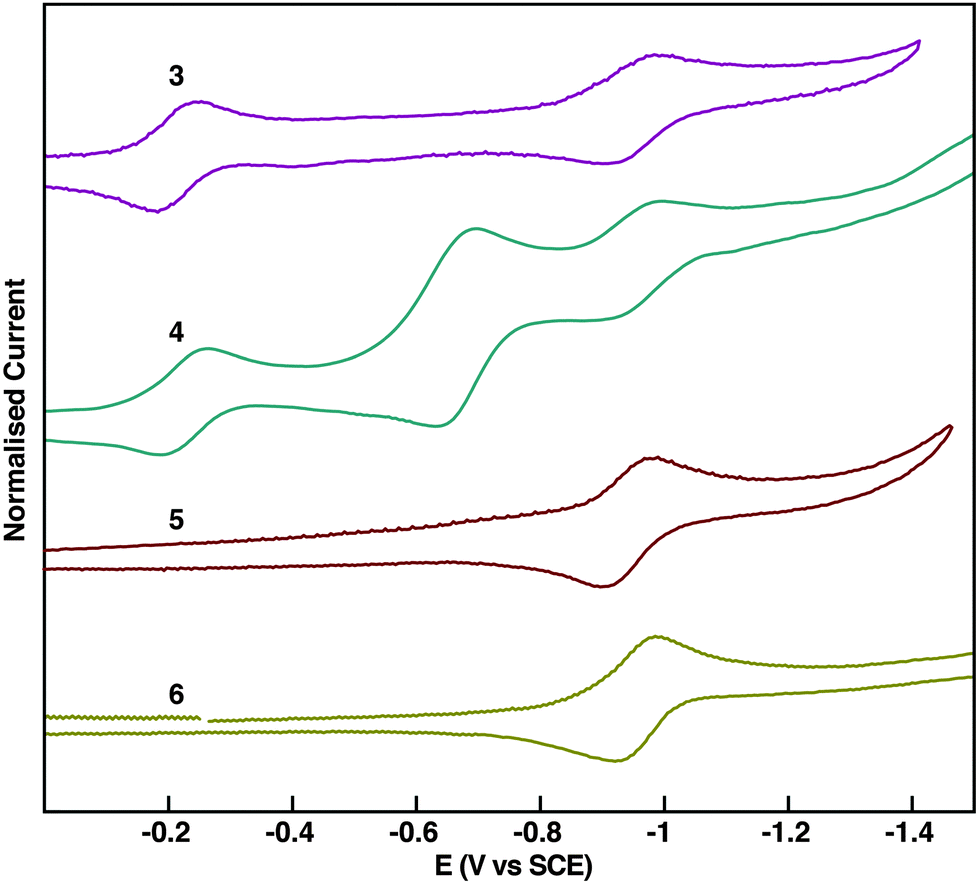 | ||
| Fig. 4 Normalised cyclic voltammograms of 3–6 in nBu4PF6/THF. | ||
The position of the energy levels was estimated from the optical and electrochemical characterisation and was correlated with DFT calculations (Table 1). The energy gaps estimated from the absorption onsets reflect that enabling the conjugation through the K region lowers the HOMO–LUMO gaps. The HOMO–LUMO gap of 3 is 0.35 eV lower than that of 4. Meanwhile, the HOMO–LUMO gap of 5 is 0.69 eV lower than that of 6. As expected, the HOMO–LUMO gap decreases when the linear conjugation is extended from one to two fused pyrazine rings. This is 0.24 eV when comparing 3 and 5, and 0.58 eV when comparing 4 and 6. The experimentally-estimated HOMO–LUMO gaps of 3–6 follow the same trends of those calculated by DFT (B3LYP-CH2Cl2-6311g+(d2,p)/B3LYP-CH2Cl2-631(d,p)) (Table S1, ESI†). The LUMO levels were estimated from the potential onsets of the first reduction waves. The ELUMO values for 3 and 4 are in the same range (−4.2 eV), which reflects the small influence of conjugation along the K region on the LUMO. Similarly, the ELUMO values for 5 and 6 are in the same range (−3.6 eV). Reasonably, the ELUMO values for 3 and 4 are substantially lowered (∼0.6 eV) in comparison to 5 and 6 because of the extended conjugation with the additional pyrazine ring. All the above indicate that the observed optoelectronic properties are indeed an effect of the destabilisation of the HOMO, while the LUMO is independent of the conjugation along the K region. This is also consistent with DFT calculations (Fig. 5), in which the shapes of the LUMO orbitals are nearly independent when comparing 3 and 5 with their parent K-truncated 4 and 6, respectively. Conversely, there are substantial differences in the shapes of the HOMO orbitals when comparing 3 and 5 with their K-truncated analogues 4 and 6, respectively. In the case of 3 and 5 the HOMOs are quite similar and spread along the PPA core but with larger orbital coefficients over the pyrene ring than on the pyrazine ring both compounds possess a large double bond character on the K region, in agreement with Clar rules.11 In the case of K-truncated 4 and 6, the shape of the HOMO differs completely from that observed in 3 and 5. The presence of the diketals suppresses the double bond character on the K region and results in a HOMO with large orbital coefficients over the lateral rings of the pyrene core. The HOMO of K-truncated 4 and 6 corresponds to the HOMO−1 of 3 and 5, which is coherent with the differences in intensities found in the simulated spectra. The experimental HOMO → LUMO transition in 3 is consistent with an intramolecular charge transfer state as the orbital coefficients in HOMO are mostly localised on the pyrene moiety, while in the LUMO they are mostly localised on the dicyanopyrazopyrazine.
| λ abs | λ em |
E
I1/2b |
E
II1/2b |
E
III1/2b |
E gap | E LUMO | |
|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2 (nm). b Measured in 0.1 M nBu4PF6/THF (V), Ag wire pseudoreference electrode referenced vs. SCE using ferrocene (Fc) as internal standard (EFc(SCE)1/2 = +0.48 V). c Estimated from absorption onset (eV). d Estimated from CV EONSET (eV) according to ELUMO = −4.8 − e(EONSET − EFc1/2) where EFc1/2 was measured in situ. | |||||||
| 3 | 571 | 687 | −0.23 | −0.95 | — | 1.85 | −4.26 |
| 4 | 445 | 500 | −0.21 | −0.65 | −0.94 | 2.20 | −4.20 |
| 5 | 455 | 538 | −0.94 | — | — | 2.09 | −3.57 |
| 6 | 390 | 514 | −0.95 | — | — | 2.78 | −3.61 |
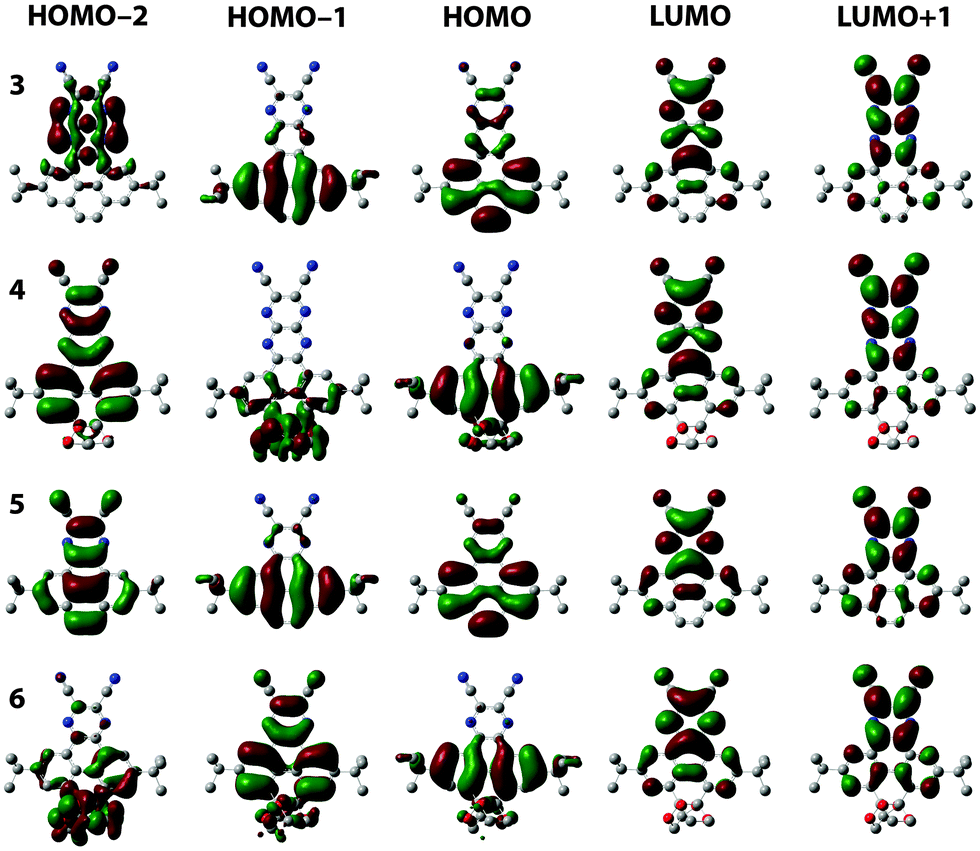 | ||
| Fig. 5 Frontier orbitals for compounds 3–6. | ||
Overall, we have shown that PPA 3 with only four linearly-fused rings emits at red to NIR wavelengths (λem = 687 nm), which is in contrast to the visible emission typically observed for PPA and n-azaacenes with an equivalent number of linearly-fused rings. This behaviour is consistent with an emissive intramolecular charge transfer state, as a result of the highly polarised electronic structure of its aromatic core and the conjugation along the K region. In fact, this combined experimental/theoretical study illustrates the influence of the conjugation along the K region on the oscillator strength of the HOMO → LUMO transition, and thus, on the overall optoelectronic properties. Nevertheless, the electron affinity is independent of the conjugation at the K region, as confirmed by cyclic voltammetry measurements, which indicate that the observed emission is indeed an effect of the destabilisation of the HOMO that is located over the pyrene residue, while the LUMO remains unaltered over the pyrazine rings. Overall, this work illustrates that it is possible to obtain PPA with enhanced properties by means of a suitable functionalization, thereby opening new perspectives for PPA in fields where NIR chromophores have already found applications.
We are grateful to the Basque Science Foundation for Science (Ikerbasque), POLYMAT, University of the Basque Country (SGIker), Deutsche Forschungsgemeinschaft (AU 373/3-1 and MA 5215/4-1), Gobierno de España (Ministerio de Economía y Competitividad, MAT2012-35826), Gobierno Vasco (BERC program), Diputación Foral de Guipuzcoa, the European Union (ERA-Chemistry, Career Integration Grant No. 618247, and FEDER), and Fundación Iberdrola (Ayudas a la Investigación en Energía y Medio Ambiente 2013-2014). MMF acknowledges support from the Portuguese “Fundação para a Ciência e a Tecnologia” and access to computing facilities financed by ON2 (NORTE-07-0162-FEDER-000086).
Notes and references
- (a) G. Tregnago, C. Fléchon, S. Choudhary, C. Gozalvez, A. Mateo-Alonso and F. Cacialli, Appl. Phys. Lett., 2014, 105, 143304 CrossRef PubMed; (b) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed; (c) B. Stender, S. F. Völker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943–2947 CrossRef CAS PubMed.
- (a) C. Y.-S. Chung, S. P.-Y. Li, M.-W. Louie, K. K.-W. Lo and V. W.-W. Yam, Chem. Sci., 2013, 4, 2453–2462 RSC; (b) T. Wang, Q.-J. Zhao, H.-G. Hu, S.-C. Yu, X. Liu, L. Liu and Q.-Y. Wu, Chem. Commun., 2012, 48, 8781–8783 RSC.
- (a) S.-Y. Lim, K.-H. Hong, D. I. Kim, H. Kwon and H.-J. Kim, J. Am. Chem. Soc., 2014, 136, 7018–7025 CrossRef CAS PubMed; (b) K. R. Bhushan, P. Misra, F. Liu, S. Mathur, R. E. Lenkinski and J. V. Frangioni, J. Am. Chem. Soc., 2008, 130, 17648–17649 CrossRef CAS PubMed; (c) J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed.
- A. Mateo-Alonso, Chem. Soc. Rev., 2014, 43, 6311–6324 RSC.
- (a) U. H. F. Bunz, Chem. – Eur. J., 2009, 15, 6780–6789 CrossRef CAS PubMed; (b) U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- (a) A. Mateo-Alonso, C. Ehli, K. H. Chen, D. M. Guldi and M. Prato, J. Phys. Chem. A, 2007, 111, 12669–12673 CrossRef CAS PubMed; (b) A. Mateo-Alonso, N. Kulisic, G. Valenti, M. Marcaccio, F. Paolucci and M. Prato, Chem. – Asian J., 2010, 5, 482–485 CrossRef CAS PubMed; (c) N. Kulisic, S. More and A. Mateo-Alonso, Chem. Commun., 2011, 47, 514–516 RSC; (d) S. More, R. Bhosale, S. Choudhary and A. Mateo-Alonso, Org. Lett., 2012, 14, 4170–4173 CrossRef CAS PubMed; (e) S. More, R. Bhosale and A. Mateo-Alonso, Chem. – Eur. J., 2014, 20, 10626–10631 CrossRef CAS PubMed; (f) S. More, S. Choudhary, A. Higelin, I. Krossing, M. Melle-Franco and A. Mateo-Alonso, Chem. Commun., 2014, 50, 1976–1979 RSC.
- S. Miao, S. M. Brombosz, P. v. R. Schleyer, J. I. Wu, S. Barlow, S. R. Marder, K. I. Hardcastle and U. H. F. Bunz, J. Am. Chem. Soc., 2008, 130, 7339–7344 CrossRef CAS PubMed.
- B. D. Lindner, Y. Zhang, S. Hofle, N. Berger, C. Teusch, M. Jesper, K. I. Hardcastle, X. Qian, U. Lemmer, A. Colsmann, U. H. F. Bunz and M. Hamburger, J. Mater. Chem. C, 2013, 1, 5718–5724 RSC.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- R. García, S. More, M. Melle-Franco and A. Mateo-Alonso, Org. Lett., 2014, 16, 6096–6099 CrossRef PubMed.
- E. Clar, The Aromatic Sextet, Wiley, London, 1972 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis, characterisation and theoretical calculations of 3–6. See DOI: 10.1039/c5cc02459e |
| This journal is © The Royal Society of Chemistry 2015 |