
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple single-crystal-to-single-crystal guest exchange in a dynamic 1D coordination polymer†
Javier
Martí-Rujas
*a,
Simone
Bonafede
b,
Dorearta
Tushi
b and
Massimo
Cametti
*b
aCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, Via Pascoli 70/3, 20133 Milano, Italy. E-mail: javier.rujas@iit.it; Fax: +392399 9866; Tel: +392399 9829
bDipartimento di Chimica Materiali e Ingegneria Chimica “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milan, Italy. E-mail: massimo.cametti@polimi.it
First published on 11th June 2015
Abstract
A novel 1D coordination polymer that dynamically expands or shrinks upon the uptake of vapours of volatile small chlorinated molecules, such as 1,2-dichloroethane (DCE), dichloromethane (DCM) and trichloromethane (TCM), is reported. This system is robust enough to withstand multiple guest exchange via single-crystal-to-single-crystal transformation, as proved by 1H-NMR and X-ray diffraction. The single crystal of guest-free, host framework, stable at 400 K, can also be obtained.
Coordination polymers having potential internal voids, often termed as metal organic frameworks (MOFs), are versatile hybrid materials, generated by the union of organic ligands and metal ions, that have shown important applications in many areas as diverse as catalysis, gas adsorption, conductivity, etc.1 This versatility stems from the vast library of organic ligands that modern synthesis can develop and, at the same time, the different coordination modes that metals can adopt. Such combination provides highly tunable and diversified materials, a crucial difference with respect to conventional porous materials like zeolites. MOFs are particular, as their frameworks can be robust yet maintain a degree of flexibility,2 allowing for dynamic structural transformations upon external stimuli.3 Such transformations have been essential to understand fundamental aspects of solid-state chemistry in general, and structure–function relationships in MOFs.4
Not surprisingly, X-ray crystallography is a central technique in such investigations, as it allows for the visualization of the three dimensional molecular arrangement at the atomic scale, particularly when guest adsorption processes occur via single-crystal-to-single-crystal (SC–SC) transformations.5 A less frequent type of reaction, which has been reported for 2D or 3D coordination polymers,6 is represented by multiple SC–SC guest exchange processes, where different guest molecules are exchanged sequentially one after the other. These transformations require robust frameworks since structural modification could be quite demanding, especially if reiterated. This explains why multiple SC–SC processes are seldom reported in coordination networks formed by 1D polymer chains, which are generally less robust compared to the 2D or 3D counterparts.7
Here, we report on a new dynamic coordination polymer synthesized by the combination of ZnCl2 with a linear non-chelating bidentate ligand (L) (Scheme 1). Its single crystal X-ray diffraction (SCXRD) characterization reveals 1D –Zn–L–Zn– chains that upon packing yield arrays of non-interpenetrated 1D channels. These channels are filled with TCM molecules (1·TCM). Notably, the same crystal can exchange TCM with DCM and DCE, via multiple SC–SC reactions whose products, 1·DCM and 1·DCE, respectively, were fully characterized by X-ray and NMR techniques.‡ This exchange is not performed by immersing the single crystals into a solution of the chosen solvent, but by directly exposing them to solvent vapours. Interestingly, solvent guest exchange is not always reversible, in that once 1·DCE is formed, exposure of its crystals to TCM or DCM vapours has no effect. This points to a marked selectivity in the solvent exchange process. Notably, the crystals are stable at room temperature, in contact with air for more than one month and are thermally stable up to 400 K.
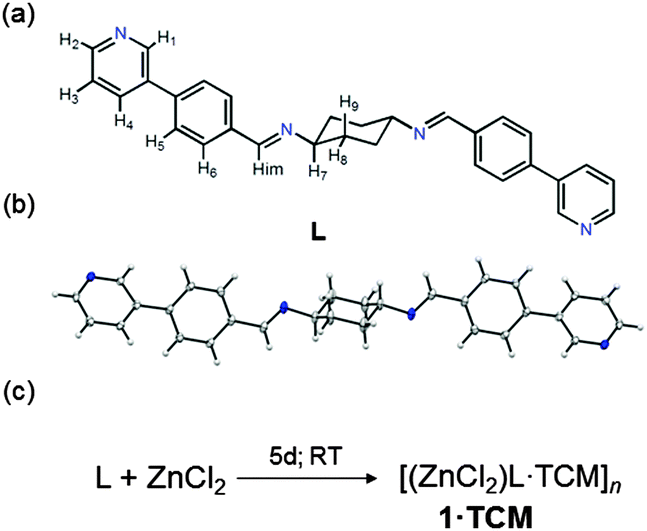 | ||
| Scheme 1 Chemical structure (a) and crystal structure (b) obtained from SCXRD of ligand L. Synthesis of the coordination polymer 1·TCM upon complexation of L with ZnCl2via slow crystallization method (c). | ||
The synthesis of L was performed by reacting 1,4-diaminocyclohexane and 4-(3-pyridinyl)benzaldehyde under refluxing conditions.8 Single crystals of 1·TCM were grown using a three layer TCM–nitrobenzene–methanol solution (ESI†).8 The 1·TCM crystal structure was fully characterized at 100 K by SCXRD.8 As shown in Fig. 1a, the Zn–N coordination results in 1D linear polymer chains which in turn pack as a consequence of the bulkiness of the central cyclohexyl moieties and the 3-pyridinyl substitution. The tendency of π–π stacking aggregation of the flat aromatic region is hampered by this structural mismatch. Weak intermolecular C–H⋯π interactions between the cyclohexane ring in one polymer chain and the pyridine rings of the adjacent polymer chains can be also detected. As a result of the relative orientations among the stacked 1D chains, a channel structure including TCM guest molecules (disordered) is formed. The channels expand infinitely along the crystallographic a-axis (Fig. 1b) and account for ca. 23.8% of the unit cell volume which corresponds to 791 Å3.9
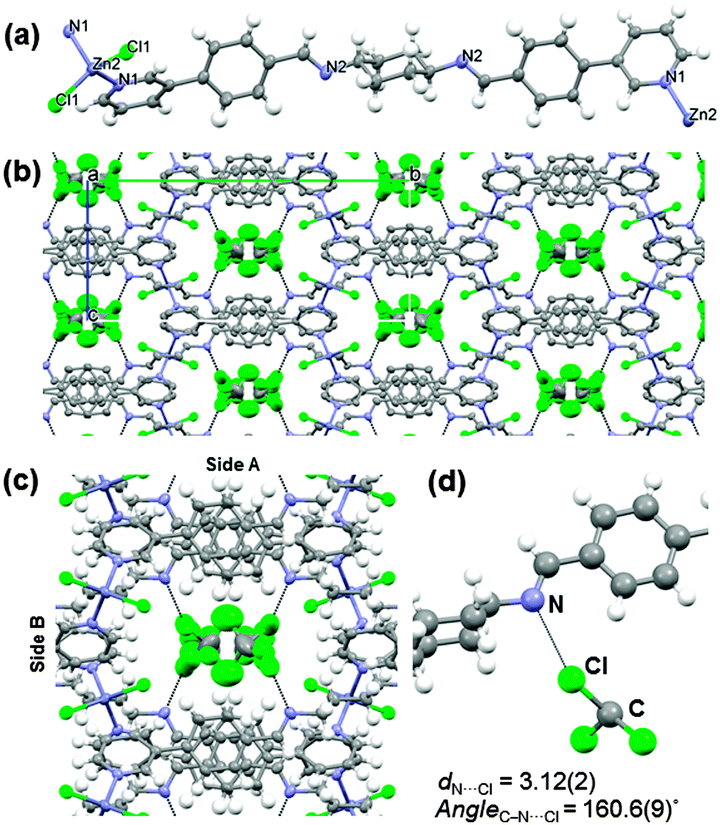 | ||
| Fig. 1 Single crystal structure of 1·TCM. (a) View of the 1D polymer chain in 1·TCM; (b) crystal packing viewed along the crystallographic a-axis showing the host guest interactions (dashed line). Hydrogen atoms have been omitted for clarity; (c) detailed view of the interior of a channel; (d) zoomed view of one of the disordered trichloromethane molecules showing the specific host–guest interactions in the channel. Colour code: carbon (gray); nitrogen (blue); chlorine (green); hydrogen (white). | ||
The internal surface of the channels exposes L's cyclohexyl rings and iminic N atoms on one side (side A), while stacked aromatic rings and the Zn-centres point their C–Hs and Zn-coordinated chloride units inwards at the other side (side B) (Fig. 1c). As a consequence of such a channel environment, N⋯Cl (d = 3.12(2) Å and C–Cl⋯N angle = 160.6(9)°) contacts between TCM and the nitrogen at the iminic bond are observed (see Fig. 1d).10 In contact with air, at room temperature, the single crystallinity of 1·TCM is maintained.
The presence of 1D channels in 1·TCM allows for guest exchange studies. Although not very common, materials of this kind were indeed used to trap different guest molecules from the gas phase via gas/solid reaction.11 We started investigating the replacement of TCM with the structurally very similar DCM. Thus, a single crystal of 1·TCM glued in a glass capillary was placed in an enclosed chamber including a vial with 1 mL of DCM. The sealed chamber containing 1·TCM and DCM was then left overnight (ca. 12 h). Once the crystal was removed from the chamber, it was observed under optical microscopy and showed no change in size, morphology and had no cracks. Then, the single crystal was mounted in the diffractometer and analysed by SCXRD at 100 K.8
Crystallographic analysis revealed that DCM replaced the original TCM guest to give the new porous coordination polymer 1·DCM. As a result of the guest exchange, a small variation in the lattice parameters (Fig. 2b) occurs, resulting in a decrease of unit cell volume whilst maintaining the space group (Pnna). After the guest exchange, the variation in the channel shape is not very significant and a similar volume space occupied by DCM (23.6% of the unit cell (775 Å3)) is observed. While in 1·TCM the solvent is disordered over two positions, DCM is found over four positions in 1·DCM.8 Importantly, the guest exchange occurs without loss of crystallinity and, therefore, via a SC–SC transformation. Due to the similarity of the two guest solvents and their considerable disorder, additional confirmation by 1H NMR is also provided (vide infra).
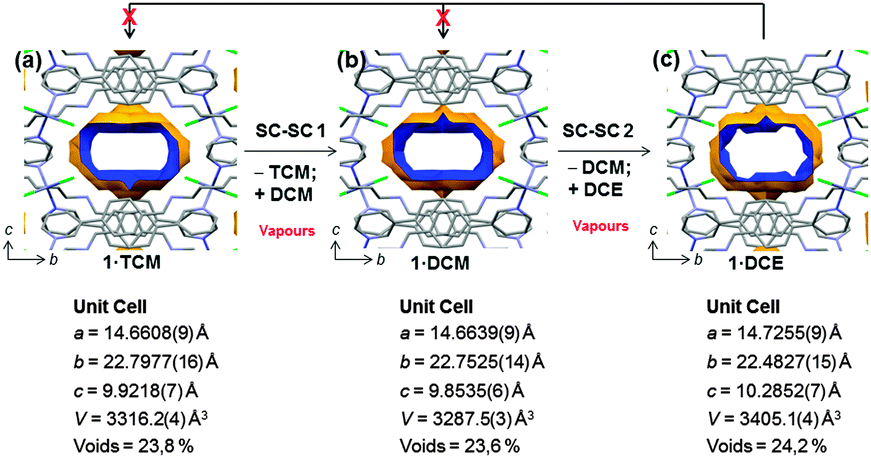 | ||
| Fig. 2 Voids corresponding to the contact surface area depicting the framework adaptability to various guest molecules via multiple SC–SC gas-solid reactions and unit cell parameters: channel view along the a-axis of as synthesized 1·TCM (a); TCM replaced by DCM (SC–SC-1) (b); DCM replaced by DCE (SC–SC-2) (c). The data were recorded at 100 K. Hydrogen atoms are omitted for clarity. Colour code as in Fig. 1. | ||
Since the crystallinity is maintained after the first SC–SC guest exchange, we attempted a second SC–SC transformation by exposing 1·DCM overnight to a third different chlorinated guest molecule: DCE. The single crystal maintained its integrity allowing complete single crystal structure elucidation after the second guest exchange process, again in a SC–SC fashion.8 SCXRD clearly shows that the incoming DCE guest molecules are actually in the channel, therefore forming the coordination polymer 1·DCE. We note that in this case, no specific host guest interactions can be detected.12 The channel shape shows a considerable change (24.2% of unit cell that corresponds to 823 Å3) when compared to 1·DCM due to an anisotropic lattice variation (Fig. 2c). DCE molecules did not escape the channels even after keeping the crystals in contact with air at room temperature for one month.
The DCE molecules are tightly packed inside the MOF as their inclusion leads to the most anisotropic structural deformation (max a and c axis, min b axis) in the framework, which also presents the largest volume. Interestingly, we also notice that the above SC–SC transformations are not always reversible, viz., by exposing 1·DCE single crystals to vapours of DCM or TCM no reversed guest exchange was observed,13 while 1·DCM can be converted back to 1·TCM by exposing it to TCM vapours. At the moment, we have no elements to exclude kinetic factors as being responsible for the observed lack of reversibility for the 1·DCE case. Also, the different vapour pressure of the three solvents, despite being remarkably different (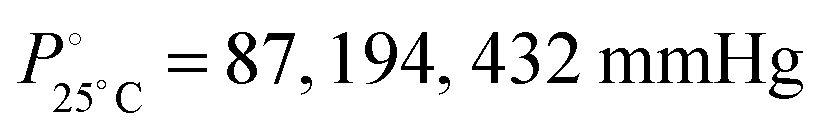 , for DCE, DCM and TCM, respectively), does not have any impact on the data interpretation since each solvent is in large excess with respect to the MOF material, in all cases, under the experimental conditions.
, for DCE, DCM and TCM, respectively), does not have any impact on the data interpretation since each solvent is in large excess with respect to the MOF material, in all cases, under the experimental conditions.
Despite the fact that the X-ray characterization of 1·TCM, 1·DCM and 1·DCE is convincing about the identity of the solvent guest present,14 we sought to gather additional confirmation by performing 1H-NMR analysis on the three species. We collected ca. 5 mg of 1·TCM, as crystallized, and about the same quantity of 1·DCM and 1·DCE after a first and second solvent guest exchange, respectively. The samples were dissolved in DMSO-d6 by heating and Fig. 3 shows the outcome of such analysis. In each case, the spectrum shows signals only of the solvent that was previously indicated by the X-ray analysis. TCM is observed in 1·TCM (Fig. 3a), but not in 1·DCM (Fig. 3b) where DCM appears, due to solvent exchange. Similarly, no DCM is present in 1·DCE, while the presence of DCE is confirmed (Fig. 3c). First of all, this rules out the possibility of an erroneous identification of the included solvent identities from the X-ray data. Secondly, this clearly confirms the occurrence of a complete solvent exchange, in accordance with the X-ray data. In all the spectra, contamination by nitrobenzene (NB) is observed, and that is due to the crystallization conditions employed. NB is a high boiling point solvent and it cannot be easily removed from the samples. Concerns about the possibility of actual inclusion of the NB evidenced by NMR into the channels can be dismissed by the fact that there is no evidence of NB from X-ray diffraction data. Moreover, attempts to include chlorobenzene, a molecule of similar size, also failed. We believe that substituted benzenes are too big to enter the MOF channels. As a reference, a spectrum of L is also shown (Fig. 3e). These above NMR analyses can also provide a quantitative evaluation of the guest solvent content of the three MOF samples, since an internal reference of known concentration, nitromethane, was added (signal at 4.41 ppm, final concentration 4.5 × 10−3 M). Data from two independent measurements indicate a solvent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio, R, equal to 0.40, 0.15 and 0.55 for the TCM, DCM and DCE, respectively. The above figures are lower than those derived from X-ray diffraction analysis which indicated R = 1 in all cases. The need to heat the sample for a complete dissolution of the material could be the reason for such discrepancy in the solvent guest content in the MOF, especially for the DCM.
:ligand ratio, R, equal to 0.40, 0.15 and 0.55 for the TCM, DCM and DCE, respectively. The above figures are lower than those derived from X-ray diffraction analysis which indicated R = 1 in all cases. The need to heat the sample for a complete dissolution of the material could be the reason for such discrepancy in the solvent guest content in the MOF, especially for the DCM.
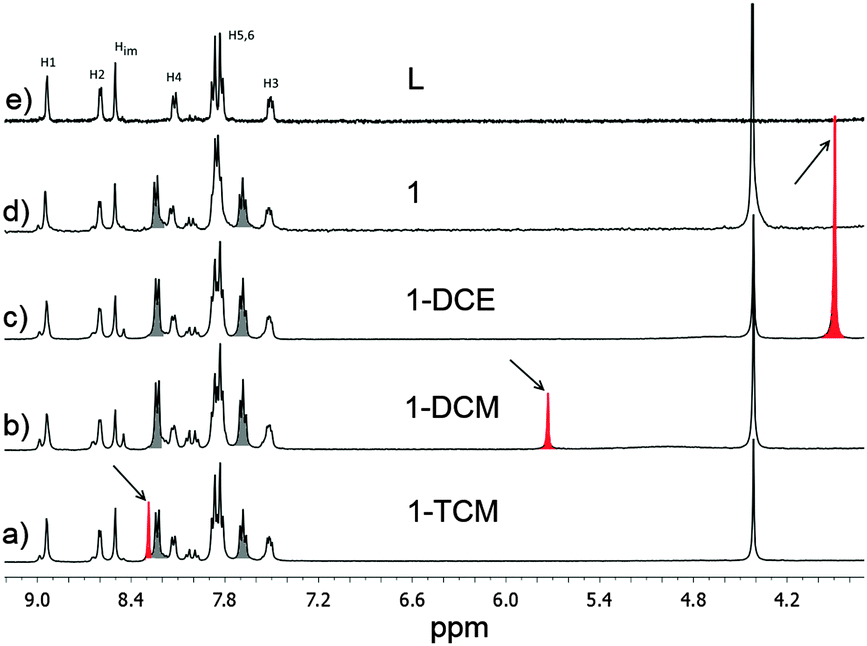 | ||
| Fig. 3 Portions of 1H-NMR (400 MHz) spectra of 1·TCM (a), 1·DCM (b), 1·DCE (c), 1 (d) and L (e) in DMSO-d6. Signal at 4.41 ppm is nitromethane used as an internal reference (4.5 × 10−3 M). The included solvents are highlighted in red, while residual nitrobenzene is in grey. | ||
A very important aspect in functional materials is represented by their stability. Thermal stability, of course, but also the ability to refrain from structural collapse upon removal of eventually trapped guest molecules. Thermo-gravimetrical analysis carried out on a microcrystalline sample of 1·TCM (vide ante) shows that complete solvent guest release is obtained at ca. 137 °C.8 Thus, the thermal stability of 1·TCM was tested by in situ heating a single crystal to 400 K overnight.15
The single crystal diffracted well at 400 K and therefore SCXRD was measured at that temperature (i.e., high temperature phase). Crystallographic analysis revealed that TCM was removed from the channels and the framework did not collapse allowing structure solution of the host framework 1 (Fig. 4a and b). We note that the changes in the lattice parameters are anisotropic and despite increasing the temperature to 400 K, the b-axis shows a length decrease of 0.28 Å. However, the channel volume after guest release is 858 Å3 which corresponds to 25% of the unit cell volume.9 The anisotropic variation in the unit cell parameters is readily observed in the shift of the diffraction peaks in the simulated XRPD pattern (Fig. 4c and d). Finally, we also analyzed by 1H-NMR a sample of 1 and, consistently, its spectrum shows no sign of TCM (Fig. 3d).
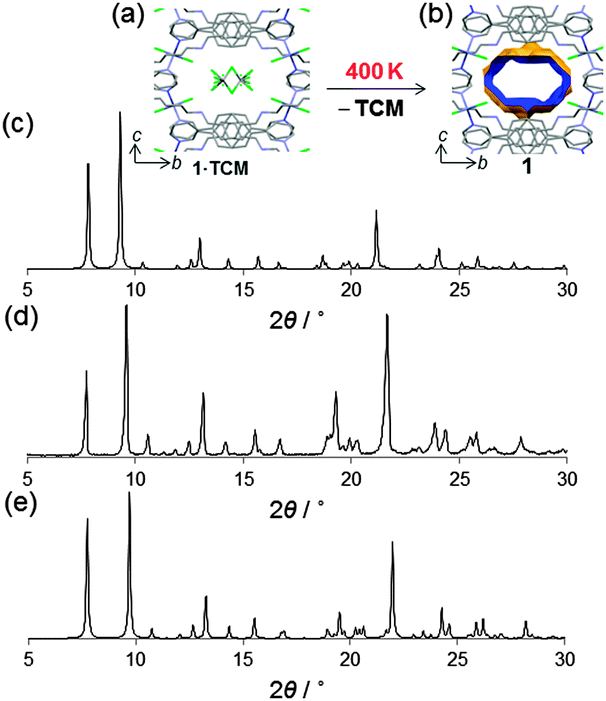 | ||
| Fig. 4 Crystal structure of the host framework 1 obtained by in situ heating 1·TCM to 400 K (a, b). Simulated XRPD pattern of 1 at 400 K (c). Experimental XRPD pattern obtained by rapidly mixing a TCM solution of L and ZnCl2 (MeOH) at 300 K (d); and simulated pattern from single crystal X-ray diffraction (100 K) 1·TCM (e). Colour code as in Fig. 1. | ||
Another important feature of this coordination polymer is that it can be produced in larger scale using fast crystallization methods.16 The white microcrystalline powder obtained shows a good agreement with the simulated powder X-ray diffraction pattern of 1·TCM (Fig. 4e).17 Therefore, 1·TCM can be easily obtained in high phase purity, quickly and in high yields as a microcrystalline powder. This is an important aspect for the realization of any industrial applications.
In conclusion, we have synthesized a new 1D coordination polymer that contains 1D channels and displays significant thermal stability. According to its guest behaviour, 1·TCM fits in Kitagawa's third type classification18 displaying breathing-like dynamic behaviour. Indeed the channel structure can dynamically adapt to various guest molecules (i.e., TCM, DCM and DCE) that can be exchanged via a multi-SC–SC reaction. While this process is more favourable in 2D and 3D linked MOFs (i.e., due to the higher number of coordination bonds), the process reported herein is noteworthy as it occurs in a 1D coordination polymer. The resulting 1·solvent species are stable and can be stored for various weeks. Importantly, our data also show preferential absorption behaviour as 1·DCE is stable in the presence of vapours of DCM and TCM at room temperature. Finally, the host framework 1 is also remarkably stable up to 400 K and, in light of the properties described above, we believe that this material could be promising for the development of a selective chemical trap for volatile organic molecules and could thus constitute a novel non-destructive remediation method to remove pollutants from the atmospheric environment.
M.C. thanks the Programma per Giovani Ricercatori “Rita Levi Montalcini” – 2009.
Notes and references
- (a) B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962 CrossRef CAS; (b) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151 CrossRef CAS; (c) S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed., 1995, 34, 2127 CrossRef CAS PubMed; (d) O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401 CrossRef CAS; (e) M. Kondo, T. Okubo, A. Asami, S. I. Noro, T. Yoshitomi, S. Kitagawa, T. Ishii, H. Matsuzaka and K. Seki, Angew. Chem., Int. Ed., 1999, 38, 140 CrossRef CAS; (f) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed; (g) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; (h) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- (a) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC; (b) C. P. Li, J. Chen, C. S. Liu and M. Du, Chem. Commun., 2015, 51, 2768 RSC; (c) C.-T. He, P.-Q. Liao, D.-D. Zhou, B.-Y. Wang, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2014, 5, 4755 RSC.
- (a) O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401 CrossRef CAS; (b) C. J. Keppert and M. J. Rosseinski, Chem. Commun., 1999, 375 RSC; (c) K. Biradha, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2000, 39, 3843 CrossRef CAS; (d) M. P. Duh, J. E. Ko and H. J. Choi, J. Am. Chem. Soc., 2002, 124, 10976 CrossRef PubMed; (e) S.-i. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2001, 124, 2568 CrossRef PubMed; (f) C. Serre, F. Millange, C. Thouvenot, M. Nogués, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef CAS PubMed; (g) S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814 CrossRef CAS PubMed; (h) K. Ohara, J. Martí-Rujas, T. Haneda, M. Kawano, D. Hashizume, F. Izumi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 3860 CrossRef CAS PubMed; (i) J. Martí-Rujas, Y. Matsushita, F. Izumi, M. Fujita and M. Kawano, Chem. Commun., 2010, 46, 6515 RSC; (j) J. Martí-Rujas, N. Islam, D. Hashizume, F. Izumi, M. Fujita and M. Kawano, J. Am. Chem. Soc., 2011, 133, 5853 CrossRef PubMed; (k) J. Martí-Rujas, N. Islam, D. Hashizume, F. Izumi, M. Fujita, H. J. Song, H. C. Choi and M. Kawano, Angew. Chem., Int. Ed., 2011, 50, 6105 CrossRef PubMed; (l) J. Martí-Rujas and M. Kawano, Acc. Chem. Res., 2013, 46, 493 CrossRef PubMed; (m) T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555 CrossRef CAS PubMed; (n) J.-K. Sun, B. Tan, L.-X. Cai, R.-P. Chen, J. Zhang and J. Zhang, Chem. – Eur. J., 2014, 20, 2488 CrossRef CAS PubMed; (o) C. Lavenn, L. Okhrimenko, N. Guillou, M. Monge, G. Ledoux, C. Dujardin, R. Chiriac, A. Fateeva and A. Demessence, J. Mater. Chem. C, 2015, 3, 4115 RSC.
- (a) M. Kawano and M. Fujita, Coord. Chem. Rev., 2007, 251, 2592 CrossRef CAS PubMed; (b) T. Kawamichi, T. Haneda, M. Kawano and M. Fujita, Nature, 2009, 461, 633 CrossRef CAS PubMed; (c) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2010, 2, 349 CrossRef PubMed; (d) J.-P. Zhang, P.-Q. Liao, H.-L. Zhou, R.-B. Lin and X.-M. Chen, Chem. Soc. Rev., 2014, 43, 5789 RSC; (e) G. K. Kole, T. Kojima, M. Kawano and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 43, 2143 CrossRef PubMed; (f) G. Mukherjee and K. Biradha, Cryst. Growth Des., 2014, 14, 3696 CrossRef CAS.
- (a) B. D. Chandler, G. D. Enright, K. A. Udachin, S. Pawsey, J. A. Ripmeester, D. T. Cramb and G. K. H. Shimizu, Nat. Mater., 2008, 7, 229 CrossRef CAS PubMed; (b) M.-S. Chen, M. Chen, S. Takamizawa, T.-A. Okamura, J. Fana and W.-Y. Sun, Chem. Commun., 2011, 47, 3787 RSC; (c) A. Demessence and J. R. Long, Chem. – Eur. J., 2010, 16, 5902 CrossRef CAS PubMed; (d) J. Xiao, Y. Wu, M. Li, B.-Y. Liu, X.-C. Huang and D. Li, Chem. – Eur. J., 2013, 19, 1891 CrossRef CAS PubMed.
- (a) S. M. Mobin, A. K. Srivastava, P. Mathur and G. K. Lahiri, Dalton Trans., 2010, 39, 8698 RSC; (b) Y.-P. Cai, X.-X. Zhou, Z.-Y. Zhou, S.-Z. Zhu, P. K. Thallapally and J. Liu, Inorg. Chem., 2009, 48, 6341 CrossRef CAS PubMed.
- For further information see the ESI†.
- The void space was calculated using a 1.2 Å probe radius: L. J. Barbour, Chem. Commun., 2006, 1163 RSC.
- Some might indicate this interaction as a rare halogen bond between a sp2 N and a Cl atom of TCM. However, we are well aware of the risk of resting such an assumption only over a simple metric analysis.
- S. Takamizawa, E. I. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4331 CrossRef CAS PubMed.
- A. Gavezzotti, CrystEngComm, 2013, 15, 4027 RSC.
- Single crystal X-ray diffraction unequivocally shows that DCE guest molecules are in the channels.
- Unit cell indexing made over several different single crystals of 1·TCM, 1·DCM and 1·DCE gave good reproducible data and each time was consistent with the inclusion of a specific guest.
- The SCXRD data set at 400 K was measured using the Oxford Cryostream device on the diffractometer. See ESI† for further details.
- At room temperature, the rapid mixing of 0.045 mmol of ZnCl2 in 1 mL of MeOH into a vigorously stirring solution of 0.045 mmol of L in 5 mL of TCM resulted in the immediate formation of a white precipitate.
- On some occasions, we have also detected another phase which has a different XRPD pattern than that of 1·TCM. So far, this phase remains unknown and we are currently investigating the conditions which can select the formation of this phase or the other, and its structural characteristics.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystal structure description, additional figures. CCDC 1055113 (L), 1055102 (1·TCM), 1055103 (1·DCM), 1055104 (1·DCE) and 1055101 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02372f |
| ‡ Single crystal data collection of L, 1·TCM, 1·DCM, 1·DCE and 1 was recorded with a Bruker X8 Prospector APEX-II/CCD diffractometer equipped with a microfocusing mirror (Cu-Kα radiation, λ = 1.54178 Å). XRPD experiments were carried out using a Bruker D2 diffractometer. |
| This journal is © The Royal Society of Chemistry 2015 |