
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular transcyclometallation: the exchange of an aryl–Pt bond for an alkyl–Pt bond via an agostic intermediate†
Paul A.
Shaw
,
Jessica M.
Phillips
,
Christopher P.
Newman
,
Guy J.
Clarkson
and
Jonathan P.
Rourke
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: j.rourke@warwick.ac.uk
First published on 10th April 2015
Abstract
Oxidation of a square-planar platinum complex leads to a five coordinate cationic intermediate that can be stabilized and trapped out via an agostic interaction with the alkyl chain of a ligand. Subsequent reaction of this species leads to the formation of an alkyl–Pt bond at the expense of an aryl–Pt bond: an intramolecular transcyclometallation.
Part of the attraction of cyclometallation, one of the oldest methods by which late transition metals can activate C–H bonds,1 is that the initial coordination directs a specific C–H bond to the metal centre, facilitating activation and providing selectivity. Cyclometallation also encompasses some less conventional2 reactions such as rollover3 reactions, and recent examples where C–H activation is preceded by reductive elimination.4
Our recent contributions to the area of cyclometallation include investigating agostic complexes of,5 C–H activation by,5a,6 and the oxidation and reduction7 of a number of cycloplatinated complexes. Some of these results, in particular the reductive coupling that occurs following oxidation, prompted us to revisit some of our earlier work with C∧N∧C pincer complexes8 and attempt to oxidise them. In these symmetrical complexes, where both carbons are formally sp2 hybridised, the possibility of reductive coupling4a is there, but as it transpires, something very different happens. The strain of the doubly cyclometallated system is relieved in a novel way, via transcyclometallation.
Transcyclometallation, originally defined9 as the exchange of one cyclometallated ligand by another, has been known for some time,10 and has been used to great effect, including its use to induce multiple11 or chiral12 cyclometallations. A more recent development has been its use to exchange one metal within the metallacycle with another, for example the exchange of gold with platinum.13 Here we present another variant: the exchange of one cyclometallated ring for another within the same complex. Furthermore, we show how it is possible to stimulate the formation of the coordinatively unsaturated intermediate that is required for this transcyclometallation reaction.
Starting from the C∧N∧C platinum DMSO complex (1a) we synthesised two different phosphine derivatives with the fourth ligand being PMe3 or PBu3 (1b and 1c respectively).
The single crystal X-ray structures of 1a and 1b are reported here for the first time, Fig. 1.
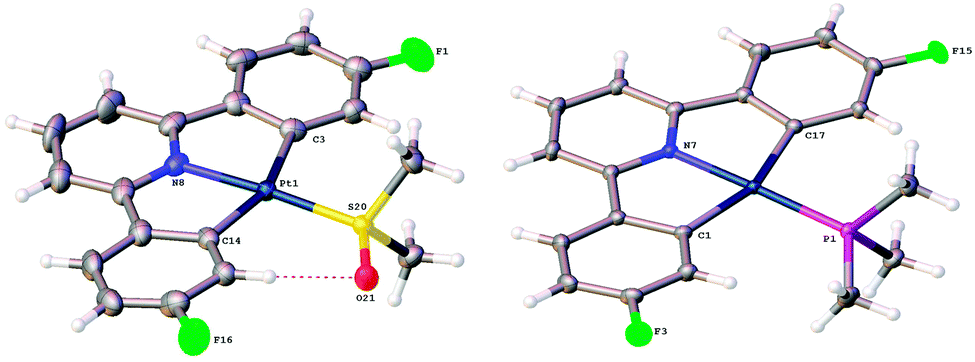 | ||
| Fig. 1 The molecular structures of 1a and 1b. Full details are in the ESI.† | ||
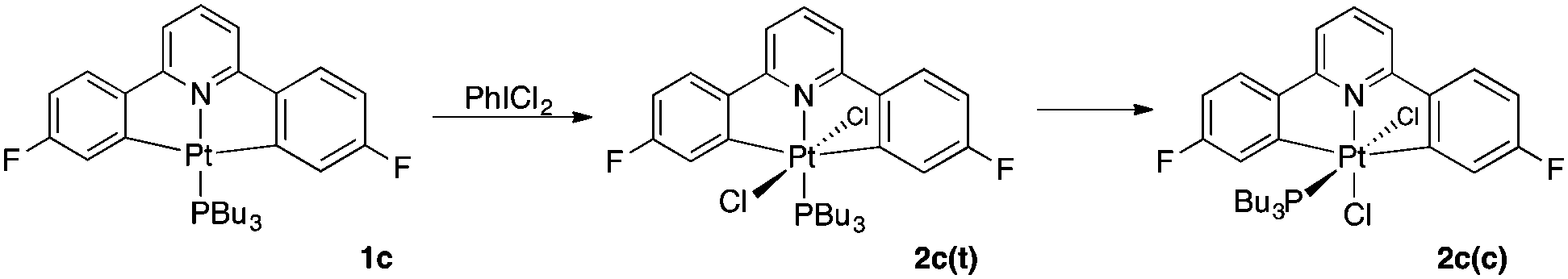
Oxidation of complexes 1b and 1c with iodobenzenedichloride is rapid, taking place in less than one minute, even at temperatures as low as −60 °C. In both cases the mechanism of oxidation appears to be the same, that is an SN2 type process,14 but there are some significant differences in the outcome of the reaction which are directly related to phosphine.
With the trimethylphosphine derivative, 1b, a rapid oxidation at low temperature gives exclusively the octahedral Pt(IV) complex with the added chlorides mutually trans, 2b(t). Upon warming the reaction mixture to room temperature, another compound is observed within 20 minutes, one that we assign as the cis product 2b(c). It would appear that the two isomers are in equilibrium, and it proved impossible to separate them, though we were able to get complete spectroscopic characterisation of the isomers from the ∼85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 (trans:cis) mixture. The PMe3 ligand is not sterically demanding, which is presumably why the cis isomer (with the phosphine over the less crowded main plane of the molecule) is not sufficiently favoured over the trans isomer to allow it to be isolated cleanly.
:15 (trans:cis) mixture. The PMe3 ligand is not sterically demanding, which is presumably why the cis isomer (with the phosphine over the less crowded main plane of the molecule) is not sufficiently favoured over the trans isomer to allow it to be isolated cleanly.

We can account for the initial formation of a trans product on the basis of a two step electrophilic oxidation of the platinum centre:14b an initial delivery of Cl+ on one face of the square planar Pt(II) centre is followed by the subsequent delivery of a Cl− to the opposite face, with no rearrangement of the existing ligands; thus the geometry of the initial product is determined by the geometry of the starting square planar platinum(II) complex. Isomerisation to relieve steric interactions takes place subsequent to the oxidation, and this pattern of behaviour is common to many of our previous studies: with DMSO ligands we observed initial formation of sulfur bound ligands which isomerised to oxygen bound ligands,5b with other complexes we observed dissociation of ligands to give agostic complexes,5c,15 and with others we observed reductive elimination.7
When the tributylphosphine complex 1c is oxidised in acetone, the reaction also cleanly gives the trans Pt(IV) product 2c(t), which again subsequently isomerises to the cis product 2c(c). Isomerism is substantially slower than for 2b(t) with complete isomerisation taking around a week in solution at room temperature.
However, both isomers could be characterised in solution and in the solid state: it proved possible to grow crystals of both 2c(c) and 2c(t), Fig. 2.
 | ||
| Fig. 2 The molecular structures of 2c(c) and 2c(t). Full details are in the ESI.† | ||
However, when the oxidation is carried out in chloroform, other products form. In the first instance, at −60 °C, another species, representing around 25% of the sample, forms; the remaining 75% is 2c(t). The new species is a complex with a symmetrical doubly cyclometallated diphenylpyridine system (one 19F resonance, with Pt satellites), with a 31P shift indicative of chelate ring formation16 (49.5 ppm compared with 2.3, −13.7 and −0.7 for 1c, 2c(t) and 2c(c), respectively) and a clear interaction of one of the end methyls of the phosphine with the platinum (satellites visible at ∼20 Hz, and a strong correlation in the 1H–195Pt spectrum). A platinum shift of −2341 indicates a Pt(IV) species. This complex is, we believe, the agostic species identified as 3 in the scheme below, and arises from the alkyl chain of the phosphine trapping out the five coordinate intermediate in an intramolecular fashion.

The agostic species is not very stable and rapidly converts to another species at above −40 °C; the new complex is reasonably stable at room temperature and indeed in air. Spectroscopic data now suggest an unsymmetrical diphenylpyridine system (two 19F resonances, one with satellites, one without), the phosphine still in a ring, though of a different size than before (31P chemical shift of 39.3), the platinum still in oxidation state +4 (195Pt shift of −2570) and a direct platinum bond to one of the alkyl groups of the phosphine. The coupling pattern on this platinated alkyl chain indicates a five membered ring (one multiplet at 2.45 ppm in the 1H NMR, relative integral 1 with platinum coupling of ∼100 Hz, coupling to a doublet, relative integral 3 with a 1H shift of 0.42 ppm and platinum satellites of 38 Hz). The presence of coupling from phosphorus to the protons in the remaining cyclometallated aryl ring suggests the P is still trans to the N and an NOE interaction suggests the Me group of the alkylated chain is positioned towards this same aryl ring. We were further able to identify the presence of an uncyclometallated phenyl ring which contains four hydrogens (two sets of two, one set of which has an agostic type interaction5c with the Pt) and we believe it to be 4 in the scheme above. In contrast to 2c(c), 2c(t) and 5 (below), recording an ESI mass spectrum on 4 gave very intense peaks that correspond to 4 as drawn, providing further evidence for the suggestion that it is an agostically stabilised cation.
Complex 4 converts to another species when we attempted to purify it by column chromatography, if it is treated with NaCl (see below), or if it is simply left in solution for more than a few hours at room temperature and we were unable to completely characterise it. We were, however, able to purify by chromatography and fully characterise this new species that 4 transforms to and the identity of this final species helps to confirm our suggestions above. All the salient features of the structure of this final complex can be deduced from the NMR data (a Pt chemical shift of −2721 indicating Pt(IV); a 31P chemical shift of 40.0 indicating one of the butyl chains is part of a cyclometallated ring, no visible coupling of this P to the protons in the cyclometallated aryl ring, suggesting the P is cis to the pyridine; two 19F resonances, one with Pt satellites, one without; an alkyl proton resonance, integral one, coupling to a methyl group and to further protons, together with a large Pt coupling; a methyl group with smaller Pt coupling; an NOE interaction indicating the close proximity in space of the single alkyl proton to the proton adjacent to Pt and F on the cyclometallated fluorophenyl ring) but it is the single crystal X-ray structure (Fig. 3) that removes any ambiguity about the connectivity of atoms. This product contains a five membered cyclometallated ring from one of the butyl chains of the phosphine, and the P donor is now cis to the N of the pyridine. Complex 5 is not cationic or agostic and has the sixth site of its octahedral geometry occupied by a further chloride. The orientation of the methyl group and the hydrogen on the platinated alkyl carbon is not disordered in the crystal structure, and the molecule as a whole is chiral; the unit cell contains equal numbers of both enantiomers.
 | ||
| Fig. 3 The molecular structure of 5. Full details are in the ESI.† | ||
The relative stereochemistry at alkyl carbon bonded to the platinum would appear to be the same in both 4 and 5, as would be expected if the rearrangement of 4 to 5 needed only a simple rotation of the cyclometallated phosphine group. The stereochemistry at this carbon makes sense if we consider the structure of 4: the cyclometallation of the alkyl chain pushes the larger methyl towards the less crowded cyclometallated aryl side of the molecule, and the smaller H towards the more congested side where the free aryl ring is.
The effect of the reaction 3 to 4 can be thought of as a transcyclometallation: both complexes have two cyclometallated rings, but whereas in 3 both are to aryl rings, in 4 one is to an aryl group and one to an alkyl chain. We have previously noted a similar type of exchange in mono-cyclometallated complexes where the steric requirements of a large t-butyl group induced strain5d,15 and we now propose it is a similar factor that is driving the reaction here: instead of the two five membered metallacycles straining and distorting the coordinating pyridine ring in 3, complex 4 has two independent five membered metallacycles without any cumulative strain, even though there has been an exchange of an aryl–Pt bond for a notionally weaker alkyl–Pt bond. The subsequent isomerisation of 4 to 5, brings the large PBu2 fragment away from a relatively congested central position to a less congested one above the plane of the diphenylpyridine, and thus also allows the final chloride ligand space to coordinate. The crystal structure of 5 provides evidence for its relatively unstrained nature: all the L–Pt–L angles are within 6° of 90°, Fig. 3. It is interesting to contrast this reactivity of 4 to that of a recent example of a platinum oxidation reaction where, instead of ultimately coordinating to the agostically stabilised metal, the final halide attacked the carbon bound to the metal, eliminating a haloalkyl group.17 Such a reaction is possible with 4 (it would result in a monocyclometallated Pt(II) species with one arm of the tributyl phosphine terminating with a CHClCH3 group) but presumably the smaller size of our ligands, together with their greater flexibility, allows the halide to coordinate directly to the metal.
As the formation of the initial agostic species and its transformation to complexes containing alkyl–Pt bonds is of considerably more interest than the simple oxidation of 1c to 2c(t), we sought to change the reaction conditions so as to favour this pathway. Unfortunately, the yield of 4 was low in all the organic solvents that we tried (chloroform, dichloromethane, toluene, methanol and nitromethane), with the best yield occurring in chloroform and the opposite extreme being toluene, acetone and nitromethane where we were unable to detect anything other than 2c(t). We can understand this on the basis of needing to prevent the combination of free chloride with a five coordinate intermediate and, of these solvents, the one that best solvates the chloride, keeping it away from the platinum, is chloroform.
We thus sought an alternative strategy, and attempted oxidations in the presence of silver salts, rationalising that, if we could (at least temporarily) restrict access to chloride, we could encourage the formation of the agostic species, leading to the alkyl activated species and so on. This strategy proved successful: initiating an oxidation reaction in chloroform at −40 °C in an NMR tube in the presence of excess AgBF4 gave greater than 50% conversion of 1c to 4. It proved necessary to destroy excess silver salt with sodium chloride to prevent extensive degradation at temperatures above −20 °C. Gratifyingly, it proved possible to completely suppress the formation of 2c(t) when the oxidation was undertaken in a conventional reaction vessel in the presence of AgBF4 with good stirring; the subsequent addition of NaCl resulted in the formation of 5 only. Silver has been observed to enhance C–H activation,18 but there is no evidence here to suggest its presence is altering the reaction course.
As another alternative synthetic route to 5, we also treated 2c(t) with AgBF4. While this reaction did not give high yields of isolable complexes, after quenching with NaCl, complex 5 could clearly be seen to be present in solution. Thus the principle of generating the 5 coordinate complex via halide abstraction from the octahedral Pt(IV) complex is valid, though we did not find it to be a practical alternative to the synthetic routes directly from 1c.
Formally five coordinate 16 electron species of platinum(IV) are frequently invoked as reaction intermediates,14a but rarely detected.17,19 Here we have demonstrated that by preventing such a species from combining with a simple chloride ligand (via the additional stabilisation that an agostic interaction offers) further avenues of reactivity are opened up. In our particular case an intramolecular transcyclometallation reaction occurs and results in the formation of an alkyl–Pt bond at the expense of an aryl–Pt bond.
We thank EPSRC for a DTG award to PAS and support from Advantage West Midlands (AWM) (part funded by the European Regional Development Fund) for the purchase of a high resolution mass spectrometer and the XRD system that was used to solve the crystal structures.
Notes and references
- M. Albrecht, Chem. Rev., 2009, 110, 576–623 CrossRef PubMed.
- I. Omae, Curr. Org. Chem., 2014, 18, 2776–2795 CrossRef CAS.
- (a) G. Minghetti, S. Stoccoro, M. A. Cinellu, B. Soro and A. Zucca, Organometallics, 2003, 22, 4770–4777 CrossRef CAS; (b) G. Minghetti, S. Stoccoro, M. A. Cinellu, G. L. Petretto and A. Zucca, Organometallics, 2008, 27, 3415–3421 CrossRef CAS.
- (a) M. Crespo, C. M. Anderson, N. Kfoury, M. Font-Bardia and T. Calvet, Organometallics, 2012, 31, 4401–4404 CrossRef CAS; (b) C. M. Anderson, M. Crespo, N. Kfoury, M. A. Weinstein and J. M. Tanski, Organometallics, 2013, 32, 4199–4207 CrossRef CAS.
- (a) S. H. Crosby, G. J. Clarkson and J. P. Rourke, J. Am. Chem. Soc., 2009, 131, 14142–14143 CrossRef CAS PubMed; (b) S. H. Crosby, G. J. Clarkson, R. J. Deeth and J. P. Rourke, Organometallics, 2010, 29, 1966–1976 CrossRef CAS; (c) S. H. Crosby, G. J. Clarkson, R. J. Deeth and J. P. Rourke, Dalton Trans., 2011, 40, 1227–1229 RSC; (d) H. R. Thomas, R. J. Deeth, G. J. Clarkson and J. P. Rourke, Organometallics, 2011, 30, 5641–5648 CrossRef CAS.
- (a) C. P. Newman, K. Casey-Green, G. J. Clarkson, G. W. V. Cave, W. Errington and J. P. Rourke, Dalton Trans., 2007, 3170–3182 RSC; (b) J. Mamtora, S. H. Crosby, C. P. Newman, G. J. Clarkson and J. P. Rourke, Organometallics, 2008, 27, 5559–5565 CrossRef CAS.
- (a) S. H. Crosby, H. R. Thomas, G. J. Clarkson and J. P. Rourke, Chem. Commun., 2012, 48, 5775–5777 RSC; (b) S. H. Crosby, G. J. Clarkson and J. P. Rourke, Organometallics, 2012, 31, 7256–7263 CrossRef CAS.
- (a) G. W. V. Cave, N. W. Alcock and J. P. Rourke, Organometallics, 1999, 18, 1801–1803 CrossRef CAS; (b) G. W. V. Cave, F. P. Fanizzi, R. J. Deeth, W. Errington and J. P. Rourke, Organometallics, 2000, 19, 1355–1364 CrossRef CAS; (c) C. P. Newman, G. W. V. Cave, M. Wong, W. Errington, N. W. Alcock and J. P. Rourke, J. Chem. Soc., Dalton Trans., 2001, 2678–2682 RSC.
- M. Albrecht, P. Dani, M. Lutz, A. L. Spek and G. v. Koten, J. Am. Chem. Soc., 2000, 122, 11822–11833 CrossRef CAS.
- (a) A. D. Ryabov and A. K. Yatsimirsky, Inorg. Chem., 1984, 23, 789–790 CrossRef CAS; (b) A. D. Ryabov, Inorg. Chem., 1987, 26, 1252–1260 CrossRef CAS.
- H. P. Dijkstra, M. Albrecht and G. van Koten, Chem. Commun., 2002, 126–127 RSC.
- F. X. Roca, M. Motevalli and C. J. Richards, J. Am. Chem. Soc., 2005, 127, 2388–2389 CrossRef CAS PubMed.
- M. Hejda, L. Dostal, R. Jambor, A. Ruzicka, R. Jirasko and J. Holecek, Eur. J. Inorg. Chem., 2012, 2578–2587 CrossRef CAS PubMed.
- (a) J. Forniés, A. Martín, R. Navarro, V. Sicilia and P. Villarroya, Organometallics, 1996, 15, 1826–1833 CrossRef; (b) L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735–1754 CrossRef CAS PubMed.
- S. H. Crosby, G. J. Clarkson and J. P. Rourke, Organometallics, 2011, 30, 3603–3609 CrossRef CAS.
- (a) L. S. Meriwether and J. R. Leto, J. Am. Chem. Soc., 1961, 83, 3192–3196 CrossRef CAS; (b) P. E. Garrou, Chem. Rev., 1981, 81, 229–266 CrossRef CAS.
- O. Rivada-Wheelaghan, M. Roselló-Merino, J. Díez, C. Maya, J. López-Serrano and S. Conejero, Organometallics, 2014, 33, 5944–5947 CrossRef CAS.
- T. L. Lohr, W. E. Piers, M. J. Sgro and M. Parvez, Dalton Trans., 2014, 43, 13858–13864 RSC.
- (a) U. Fekl, W. Kaminsky and K. I. Goldberg, J. Am. Chem. Soc., 2001, 123, 6423–6424 CrossRef CAS; (b) E. Khaskin, P. Y. Zavalij and A. N. Vedernikov, Angew. Chem., Int. Ed., 2007, 46, 6309–6312 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details including X-ray structures and CIF files for 1a, 1b, 2c(t), 2c(c) and 5. CCDC 1055262–1055266. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02355f |
| This journal is © The Royal Society of Chemistry 2015 |