
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chloride triggered reversible switching from a metallosupramolecular [Pd2L4]4+ cage to a [Pd2L2Cl4] metallo-macrocycle with release of endo- and exo-hedrally bound guests†
Dan
Preston
,
Alyssa
Fox-Charles
,
Warrick K. C.
Lo
and
James D.
Crowley
*
Department of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz; Fax: +64 3 479 7906; Tel: +64 3 479 7731
First published on 23rd April 2015
Abstract
A metallosupramolecular [Pd2L4]4+ cage can be cleanly converted into a [Pd2L2Cl4] metallo-macrocycle upon addition of chloride ions. The process is reversible, treatment of the [Pd2L2Cl4] macrocycle with silver(I) ions regenerates the [Pd2L4]4+ cage. Additionally, it is shown that guest molecules could be released on chloride triggered cage dis-assembly and taken up anew on re-assembly.
The ready synthesis1 and molecular recognition properties of metallosupramolecular assemblies have resulted in these systems being examined for a variety of potential applications.2 The host–guest properties of these metallosupramolecular assemblies have been exploited for a variety of proposes, including molecular storage of reactive species,3 the binding of environmental pollutants,4 catalysis,5 and the delivery of biologically relevant molecules including drugs.6 The ability of metallosupramolecular assemblies to selectively bind guest molecules within their internal cavities is clearly extremely important for these applications. However, just as useful as guest encapsulation, but harder to attain, is the ability to controllably release the guest from the cavity when required. There are two different approaches to induce guest release from metallosupramolecular assemblies: (1) alteration of the guest and its binding affinity for the host (Fig. 1a), or (2) modification of the host and its binding affinity for the guest (Fig. 1b–d). Examples of both approaches have been demonstrated previously. A number of groups have shown that neutral guest molecules will be expelled from cationic host cages upon introduction of positive charge onto the guest, either by protonation or oxidation.6a,7 Others have shown that light switchable guest molecules can be released from a cationic host cage upon UV irradiation.8 In these systems the properties of the guest are altered by some stimulus while the host remains unchanged (Fig. 1a). An alternative method is to modify the host and therefore its binding ability while leaving the guest molecule unchanged (Fig. 1b–d). There are number of ways in which this can be achieved. Yam and co-workers9 have shown that Pt-containing guest molecules can be released from [Pt2L2]2+ metallo-rectangles by protonation of the endotopic pyridine units that line walls of the cavity of the assembly. Clever and co-workers have developed a [Pd2L4]4+ cage that binds a [B12F12]2− anion. Irradiation of the host–guest adduct with UV (365 nm) induces an isomerization of the cage's ligands and expels the guest from the host cavity.10 Transmetallation has also been exploited to trigger guest release. Addition of Cu(I) ions to a Cd(II) M4L2 assembly leads to the release of encapsulated anionic croconate guest molecules.11 In these examples the metallosupramolecular assemblies' overall three-dimensional connectivity is retained but the stimulus alters the host's binding ability and leads to release of the guest molecule (Fig. 1b).
 | ||
| Fig. 1 Generic cartoon representations of stimuli-responsive guest release from metallosupramolecular assemblies; (a) stimuli induced alteration of the guest, (b) stimuli induced alteration of the host binding affinity for the guest with retention of the host's three-dimensional structure, (c) complete dis-assembly of the host architecture (d) partial dis-assembly of the host structure. | ||
Guest release can also be achieved by exploiting the dynamic nature of the metal–ligand interaction to either completely (Fig. 1c) or partially (Fig. 1d) dis-assemble the metallosupramolecular host. Nitschke and co-workers have developed a family of tetrahedral [Fe4L6]8+ cages which can bind a variety of neutral guest molecules. Treatment of these cages with either acid or tris(2-ethylamino)amine leads to the complete dis-assembly of the cages, releasing encapsulated cyclohexane12 or SF64 guest molecules. Others have developed systems that operate in a similar fashion. Yoshizawa and co-workers synthesised a [Ag2L2]2+ metallo-macrocycle from an anthracene-containing dipyridyl ligand and showed that it can strongly bind the fullerene C60.13 UV irradiation of the [C60⊂Ag2L2]2+ host–guest adduct causes the reduction of the Ag(I) ions to Ag(0) leading to the disintegration of the metallo-macrocycle and release of the encapsulated C60. Using the same anthracene-containing dipyridyl ligand Yoshizawa and co-workers generated a [Hg2L4]4+ cage system.14 Like the Ag(I) metallo-macrocycle the [Hg2L4]4+ cage is capable of encapsulating C60 or C70 fullerenes. Addition of two equivalents of Hg2+ causes the [C60⊂Hg2L4]4+ cage to rearrange to form two [Hg2L2]4+ metallo-macrocyles and release of the fullerene guest. Reformation of the [Hg2L4]4+ cage and re-binding of the fullerene was achieved through addition of two equivalents of free ligand to the metallo-macrocycle fullerene mixture.
As part of our interest in functional metallosupramolecular assemblies15 we have developed a [Pd2L4]4+ cage system, utilising ‘banana-shaped’ 2,6-bis(pyridin-3-ylethynyl)pyridine ligands, which can bind the anti-cancer drug cisplatin.16 We have shown previously, that the cage can be completely dis-assembled using an excess of 4-(N,N-dimethylamino)pyridine (DMAP) or chloride (Cl−) to release the cisplatin guest. We herein report the formation of a new, more soluble, [Pd2L4]4+ cage using a diglyme substituted tripyridyl ligand, L. This new system can be reversibly toggled between the cage and its corresponding [Pd2L2Cl4] metallo-macrocycle by the sequential addition of tetrabutylammonium chloride ([NBu4]Cl) and AgBF4 as stimuli. Furthermore, it is demonstrated that the cage can interact both endo- and exo-hedrally with guest molecules which can be released from the cage (Fig. 2a) upon treatment with chloride.
 | ||
| Fig. 2 (a) A cartoon showing the chloride induced conversion of a [Pd2L4](BF4)4 (C) into a metallo-macrocycle [Pd2L2Cl4] (M) with release of both endo- and exo-hedrally bound guest molecules; (b) the molecular structures of the ligand (L), cage (C) and metallo-macrocycle (M). The scheme (b) shows the formation of C from L and [Pd(CH3CN)4](BF4)4 and the reversible formation of M using [NBu4]Cl and AgBF4 as stimuli. Colours: carbon, grey; chloride, green; nitrogen, blue; oxygen, red; palladium, magenta. Hydrogen atoms, counterions and solvent molecules were omitted for clarity. | ||
The ligand (L, Fig. 2b), substituted with diglyme chains on the terminal pyridine rings for improved solubility, was synthesised in good yield (71%) using standard procedures (ESI†).17 Addition of [Pd(CH3CN)4](BF4)2 (1 eq.) to an acetonitrile (CH3CN) solution of L (2 eq.) gave the cage, [Pd2L4](BF4)4 (C) in good yield (Fig. 2b and ESI†). The identities of the ligand and the cage were confirmed by 1H, 13C and DOSY NMR and IR spectroscopies and high resolution electrospray mass spectrometry (HR-ESMS) (Fig. 3 and ESI†). The 1H NMR spectrum of C (Fig. 3b and ESI†) showed a single set of peaks, with the proton resonances of the terminal pyridine (Hc–e) shifted downfield relative to L (Fig. 3a and b and ESI†), consistent with complexation of L to the palladium(II) ions. Further solution phase evidence for the formation of the desired discrete cage architecture was obtained from the 1H DOSY NMR of L and C. Each of the proton signals in the individual spectra show the same diffusion coefficients (DL = 4.59 × 10−10 m2 s−1, DC = 2.16 × 10−10 m2 s−1), indicating that there is only one species present in each solution (ESI†). The ratio of the diffusion coefficients of the ligand and palladium complex in d7-dimethyformamide (DMF) was approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, consistent with the formation of a larger molecular cage species. Mass spectra (HR-ESMS) of the cage C in DMF–CH3CN solution displayed a series of isotopically resolved peaks due to [Pd2L4](4−n)+ (BF4)n (where n = 1–3) ions along with peaks due to fragmentation of the cage structure (ESI†). Additionally, the molecular structures of the ligand (L) and the cage (C) were determined unequivocally by X-ray diffraction (Fig. 2b and ESI†).
:1, consistent with the formation of a larger molecular cage species. Mass spectra (HR-ESMS) of the cage C in DMF–CH3CN solution displayed a series of isotopically resolved peaks due to [Pd2L4](4−n)+ (BF4)n (where n = 1–3) ions along with peaks due to fragmentation of the cage structure (ESI†). Additionally, the molecular structures of the ligand (L) and the cage (C) were determined unequivocally by X-ray diffraction (Fig. 2b and ESI†).
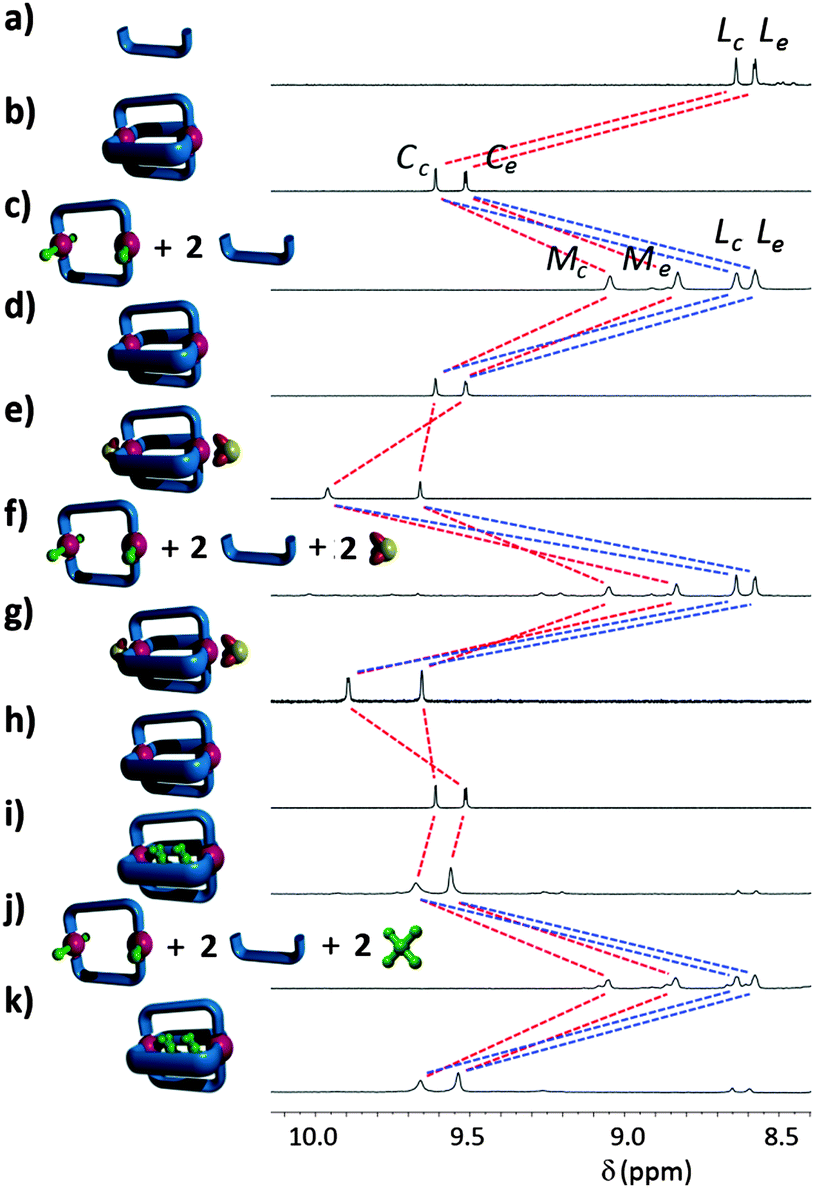 | ||
| Fig. 3 Stacked, partial 1H NMR spectra (500 MHz, d7-DMF, 298 K) showing (a) free ligand L, (b) cage C, (c) addition of [NBu4]Cl (4 eq.) giving macrocycle M and ligand L, (d) addition of AgBF4 (4 eq.) giving C, (e) addition of [NBu4](OMs) (2 eq.), (f) addition of [NBu4]Cl (4 eq.) giving M and L, and liberated MsO− anions, (g) addition of AgBF4 (4 eq.) leading to reformation of host–guest adduct [(DMF)2(MsO−)2⊂C]2+, (h) C, (i) addition of cisplatin (excess), giving [(cisplatin)2⊂C]4+ host–guest adduct, (j) addition of [NBu4]Cl (4 eq.) giving M, L, and liberated cisplatin molecules, and (k) addition of AgBF4 (4 eq.) resulting in the reformation of the host–guest adduct [(cisplatin)2⊂C]4+. | ||
Addition of four equivalents of tetrabutylammonium chloride to a d7-DMF solution (0.75 mM) of the cage C resulted in the formation of new two species (Fig. 3c). The chemical shifts and diffusion coefficient for one of the two species in solution were identical to those of the ligand L, suggesting that chloride ions were displacing some of the ligands L from the palladium(II) cage complex forming a mixture of free L and a new neutral metallo-macrocycle [Pd2L2Cl4] (M). The proton chemical shifts and diffusion coefficient (DM = 3.10 × 10−10 m2 s−1) for the new species were intermediate between those of C and L, consistent with the formation of the neutral, chloride-containing complex M. Vapour diffusion of diethyl ether into a DMF solution containing a mixture of M and L provided X-ray quality crystals of the new palladium complex unambiguously confirming the formation of the neutral metallo-macrocycle architecture (Fig. 2b and ESI†).18
Interestingly, efforts to prepare M independently by reacting [Pd(CH3CN)2Cl2] and L resulted in only insoluble polymeric products indicating that the preorganisation provided by the cage C was essential for the clean generation of the metallo-macrocycle. Treatment of the d7-DMF solution (0.75 mM) of M and L with four equivalents of AgBF4 abstracted the chloride ions from M and led to the reformation of the [Pd2L4](BF4)4 cage (C). Thus we have generated a stimuli-responsive system that can be toggled cleanly between two different metallosupramolecular architectures, a cage and a metallo-macrocycle, by the addition or removal of chloride ions.
We have previously shown that related dipalladium cages are able to bind both neutral cisplatin16,19 and anionic20 mesylate (MsO−) guest molecules.211H NMR spectra of mixtures of the new diglyme substituted cage and these guest molecules indicated that C retains the host–guest capability of the previously reported palladium(II) cage systems and bind both cisplatin and mesylate (Fig. 3).
Mixing cisplatin (excess) and [Pd2L4](BF4)4 in d7-DMF solution results in a downfield shift and broadening of the proton resonance Hc of the cage NMR (Fig. 3h and i, Δδ = 0.05 ppm), indicative of cisplatin encapsulation as we have observed previously.14,16 In d7-DMF the shift is smaller than those observed in CD3CN solution14,16 indicating that the interaction is weaker. 1H NMR titrations showed that the interaction between C and cisplatin, in the highly competitive DMF solvent, is extremely weak (K1 = 2 ± 1 M−1, K2 = 5 ± 2 M−1, ESI†).
Similar NMR experiments confirmed the interaction of mesylate anions with C. A 1H NMR mole-ratio22 titration of C with the mesylate guest indicated that two MsO− guests bind to the cage (ESI†). Addition of tetrabutylammonium mesylate (2 eq.) to a d7-DMF solution of the cage C caused downfield shifts of the proton resonances Hc and He. The shift of proton He (Δδ = 0.45 ppm) was the largest (Fig. 3e), suggesting that the MsO− anions are binding on the exo-hedral face(s) of C. The association constants (K1 = 1000 ± 100 M−1, K2 = 180 ± 20 M−1), for the interaction of the mesylate anions with C were determined using 1H NMR titration (ESI†), the values are similar to those observed in related host–guest systems.21 Slow vapour diffusion of diethyl ether into a DMF solution containing C and ten equivalents of tetrabutylammonium mesylate resulted in X-ray quality crystals of the host–guest adduct. The molecular structure (ESI†) confirmed that two mesylate anions bind on the two exo-hedral faces of the dipalladium complex and two DMF molecules fill the central cavity of the cage giving a [(DMF)2(MsO−)2⊂C]2+ host–guest adduct.
This interesting behaviour allows the possibility of C binding two cisplatin and two mesylate anions at the same time. Pleasingly the 1H NMR spectrum (ESI†) of a d7-DMF solution containing cisplatin (excess), tetrabutylammonium mesylate (2 eq.) and [Pd2L4]4+ was consistent with C interacting all four guest molecules simultaneously; two cisplatins bound in the central cavity and two mesylate anions binding on the exo-hedral faces of the cage.
Having confirmed the guest binding abilities of C we set out to see if the chloride induced structural rearrangement to the metallo-macrocycle M would lead to guest release. Addition of tetrabutylammonium chloride (4 eq.) to d7-DMF solutions containing one of the host–guest adducts (either [(cisplatin)2⊂C]4+, [(DMF)2(MsO−)2⊂C]2+ or [(cisplatin)2(MsO−)2⊂C]2+) resulted in essentially identical 1H NMR spectra (Fig. 3f and j and ESI†). The proton chemical shifts and diffusion coefficient of M and L in these spectra were identical to those observed when C was reacted with tetrabutylammonium chloride (Fig. 3c) indicating that the metallo-macrocycle M and the ligands L do not interact with either neutral cisplatin or anionic mesylate guests. Thus chloride triggered formation of M is accompanied by guest release (either two cisplatin, two mesylates or all four guests).23 Presumably the mesylate anions do not interact with the metallo-macrocycle M because this neutral complex lacks the strong cation–anion interactions that are present in the [(DMF)2(MsO−)2⊂C]2+ host–guest adduct. The lack of interaction of cisplatin with M can potentially be ascribed to the trans orientation of the central pyridyl hydrogen bond donors of the metallo-macrocycle. This new orientation means that they are no longer able to interact with both of cis-ammine ligands of cisplatin in a chelating fashion. Addition of AgBF4 (4 eq.) to these mixtures of M, L and guest molecules resulted in the reformation of the cage C and restored the host–guest interactions (Fig. 3g and k).
In conclusion we have developed a new diglyme substituted [Pd2L4]4+ cage which can be cleanly converted into [Pd2L2Cl4] metallo-macrocycle upon the addition of four equivalents of chloride. The initial [Pd2L4]4+ cage can be quantitatively reformed by treating the mixture of [Pd2L2Cl4] and 2L with Ag(I) ions. The cage showed the capacity to bind two cisplatin molecules within the internal cavity of the architecture and two mesylate anions on the exterior face, or all four guests simultaneously. Additionally, it was demonstrated that the guest molecules, bound either endo- (within the cage cavity) or exo-hedrally on the exterior face of the Pd(II) cage, were released into solution on chloride triggered cage dis-assembly and taken up anew on re-assembly.
As Pd(II)-based systems24 are one of the most common classes of metallosupramolecular architectures we are now examining if this chloride triggered cage dis-assembly can be applied to other larger Pd(II)-containing assemblies. The ability to controllably release or recall guests from metallosupramolecular architectures has potential in wide-ranging applications such as drug delivery, catalysis and environmental remediation.
This work was supported by an Otago Medical Research Foundation Laurenson Award (LA307). The authors thank Department of Chemistry, University of Otago for additional funding. DP and WKCL thank the University of Otago for PhD scholarships. DP thanks Otago Medical Research Foundation for a McQueen Summer Studentship.
Notes and references
- For some selected recent reviews see; (a) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC; (b) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (c) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (d) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- (a) B. Therrien, CrystEngComm, 2015, 17, 484–491 RSC; (b) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (c) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC; (d) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed; (b) M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CAS.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC.
- (a) Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS PubMed; (b) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- (a) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625–631 RSC; (b) Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC; (c) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Suss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- (a) J. D. Crowley, A. J. Goshe, I. M. Steele and B. Bosnich, Chem. – Eur. J., 2004, 10, 1944–1955 CrossRef CAS PubMed; (b) W.-Y. Sun, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 2002, 124, 11570–11571 CrossRef CAS PubMed; (c) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561–8562 CrossRef CAS; (d) M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039–7046 CrossRef CAS PubMed.
- G. H. Clever, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS PubMed.
- A. K.-W. Chan, W. H. Lam, Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 690–695 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- Q. Gan, T. K. Ronson, D. A. Vosburg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 1770–1773 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- (a) S. V. Kumar, W. K. C. Lo, H. J. L. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1–6 CrossRef CAS PubMed; (b) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC; (c) S. K. Vellas, J. E. M. Lewis, M. Shankar, A. Sagatova, J. D. A. Tyndall, B. C. Monk, C. M. Fitchett, L. R. Hanton and J. D. Crowley, Molecules, 2013, 18, 6383–6407 CrossRef CAS PubMed; (d) S. O. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117–12124 RSC; (e) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC; (f) J. D. Crowley, I. M. Steele and B. Bosnich, Eur. J. Inorg. Chem., 2005, 3907–3917 CrossRef CAS PubMed; (g) J. D. Crowley, A. J. Goshe and B. Bosnich, Chem. Commun., 2003, 2824–2825 RSC.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- (a) V. Gudipati, D. P. Curran and C. S. Wilcox, J. Org. Chem., 2006, 71, 3599–3607 CrossRef CAS PubMed; (b) K. J. Kilpin, M. L. Gower, S. G. Telfer, G. B. Jameson and J. D. Crowley, Inorg. Chem., 2011, 50, 1123–1134 CrossRef CAS PubMed.
- Recently, Clever and co-workers have shown that halide ions can trigger the conversion of [Pd2L4]4+ cage into a triply catenated link structure see; R. Zhu, J. Luebben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC; (b) J. E. M. Lewis, C. J. McAdam, M. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398–3400 RSC.
- J. E. M. Lewis and J. D. Crowley, Supramol. Chem., 2014, 26, 173–181 CrossRef CAS PubMed.
- A wide range of sulfonate anions have been shown to interact with Pd(II) metallo-cage architectures see; (a) G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef CAS PubMed; (b) G. H. Clever and M. Shionoya, Chem. – Eur. J., 2010, 16, 11792–11796 CrossRef CAS PubMed; (c) M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 706–710 CrossRef CAS PubMed; (d) R. Sekiya and R. Kuroda, Chem. Commun., 2011, 47, 12346–12348 RSC; (e) R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed; (f) N. J. Cookson, J. J. Henkelis, R. J. Ansell, C. W. G. Fishwick, M. J. Hardie and J. Fisher, Dalton Trans., 2014, 43, 5657–5661 RSC.
- A. S. Meyer, Jr. and G. H. Ayres, J. Am. Chem. Soc., 1957, 79, 49–53 CrossRef.
- During the course of our work, Clever and co-workers have shown that halide ions can trigger guest exchange in a double cage system see; S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef PubMed.
- N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C and DOSY NMR, HR-ESMS, molecular models and crystallographic data. CCDC 1053822–1053825. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02226f |
| This journal is © The Royal Society of Chemistry 2015 |