
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of the methylsulfinyl radical [CH3(O)S˙] with oxygen (3O2) in solid argon†
Hans Peter
Reisenauer
*a,
Jarosław
Romański
b,
Grzegorz
Mlostoń
*b and
Peter R.
Schreiner
 a
a
aInstitute of Organic Chemistry, Justus-Liebig University, Heinrich-Buff-Ring 58, D-35392 Giessen, Germany. E-mail: reisenauer@org.chemie.uni-giessen.de; Fax: +49 641-9934309; Tel: +49 641-9934380
bDepartment of Organic and Applied Chemistry, University of Lodz, Tamka 12, 91-493 Lodz, Poland. E-mail: gmloston@uni.lodz.pl; Fax: +48 42 6655162; Tel: +48 42 6355761
First published on 18th May 2015
Abstract
The atmospherically highly relevant methylsulfinyl radical (CH3(O)S˙) readily reacts with molecular triplet oxygen in cryogenic argon matrices containing small amounts of 3O2. Comparison of experimental and computed IR- and UV/Vis spectra, including isotope exchange, show that the initially formed 3O2 adduct has the structure of a peroxyl radical (CH3(O)SOO˙), which upon irradiation with UV light isomerizes to the methylsulfonoxyl radical (CH3SO3˙). The latter transforms into the methansulfonic acid radical (˙CH2SO3H) by irradiation with visible light. During the matrix photolysis small amounts of SO3 and the methyl radical were detected indicating competitive direct photodissociation.
Volatile sulfur compounds such as hydrogen sulfide and dimethylsulfide (Me2S and DMS) produced from natural plankton are emitted into the atmosphere and are converted into a plethora of sulfur products via radical processes. As part of the so-called ‘sulfur cycle’ they are of great importance for the atmospheric sulfur balance1 with DMS as the major component of biogenic emissions.2 The impact of sulfur compounds on the climate is explained based on the ‘CLAW’ hypothesis.3 DMS and other volatile sulfur compounds undergo cascade conversions in the atmosphere eventually leading to sulfuric acid and methanesulfonic acid (Scheme 1).3c The methylsulfinyl radical (1) and the methylsulfonyl radical (2) are recognized as important intermediates involved in these processes.4
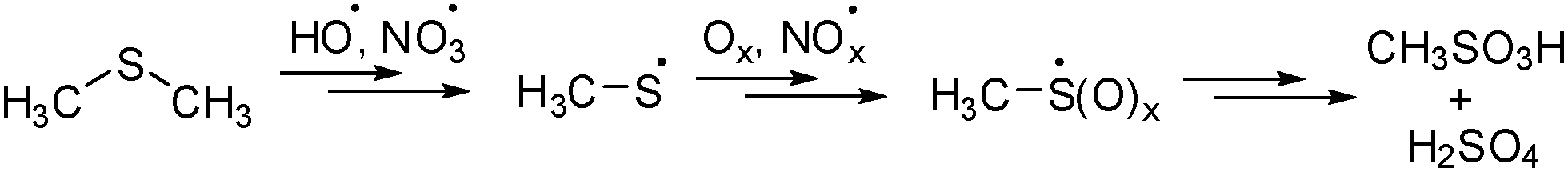 | ||
| Scheme 1 Postulated reaction cascade for dimethyl sulfide oxidation to sulfuric acid and methylsulfonic acid in the atmosphere. | ||
A recent study reports the high vacuum flash pyrolysis (HVFP) of allylmethylsulfoxide (3) generating the highly stabilized π-radical 1 isolated in an Ar matrix at 10 K.5 Similarly, 2 was trapped starting with allylmethylsulfone (4) (Scheme 2).6
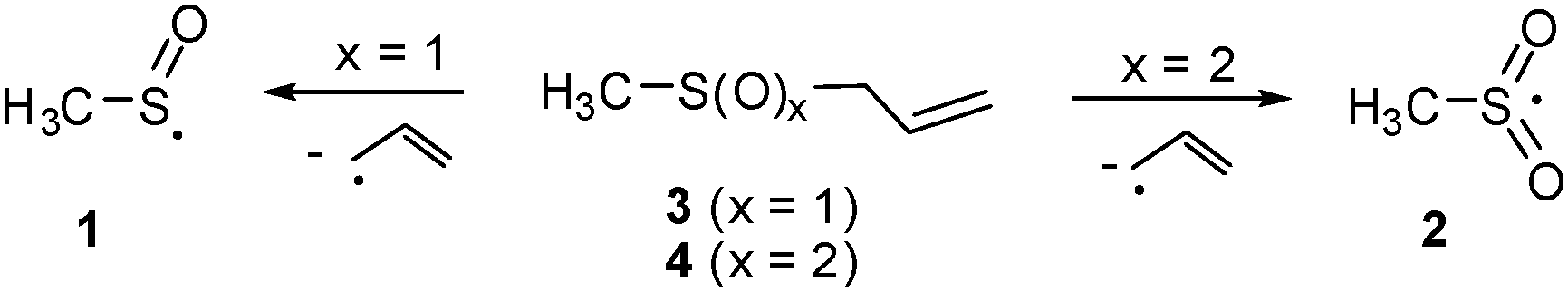 | ||
| Scheme 2 Generation of 1 and 2 starting with allylmethylsulfoxide (3) and allylmethylsulfone (4), respectively. | ||
The study showed that 2 in comparison with 1 is significantly less stable and easily undergoes partial decomposition in the gas phase leading to the formation of SO2 and the methyl radical. To date little is known about the reactions of 1 and there are only a few kinetic studies of its reactions with O2, O3, and NO2.7 In the case of the reaction with 3O2, the formation of an adduct with the structure of the methylsulfinylperoxyl radical (5) as the key intermediate was postulated (Scheme 3).8 However, to the best of our knowledge, no spectroscopic data for 5 have been presented to date.
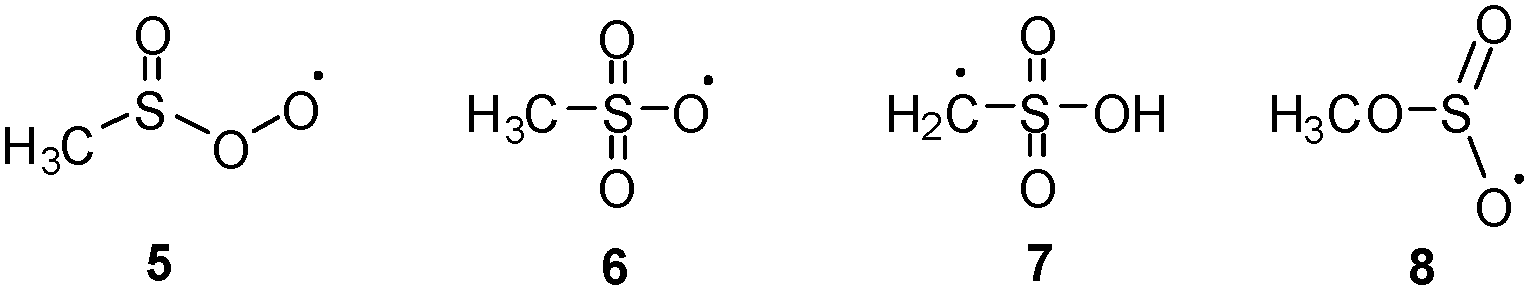 | ||
| Scheme 3 Most relevant [CH3SO3]˙ radical species 5–8. | ||
The fundamental question whether the formation of 5 by addition of 1 to molecular oxygen is an exo- or an endothermic process has not yet been unambiguously answered by theory. The ΔH° values computed at ab initio and density functional theory (DFT) levels with large basis sets vary considerably; G2(MP2) computations indicate slightly exothermic (−2.5 kcal mol−1) formation of 5.9
The main goal of the present work was to determine if this reaction takes place at all and to examine the structure of the product formed. Equally important for the understanding of atmospheric chemistry is an investigation of the photochemistry of the initially formed oxygen adduct. The matrix isolation technique currently is the best method to provide reliable answers to both questions.
Following the previously described protocol for the generation of 1,3 a sample of the precursor 3 was pyrolized at 600 °C and the stream of products was co-condensed at 10 K with a large excess of argon containing ca. 0.5, 1.0 or 5.0 vol% of oxygen, respectively (Scheme 4). In experiments with 0.5 and 1.0 vol% of 3O2 only partial conversion of 1 and the allyl radical (9) was observed. In the IR spectra the characteristic absorptions located at 800 cm−1 (for 6) and at 1068 cm−1 (for 1) were still present. In addition a new, intense band found at 1191 cm−1 provided evidence for the formation of a new product. However, in the case of experiments performed with 5 vol% 3O2 content, the conversions were complete and none of the bands of 1 or 9 were found in the pyrolysate. Two new sets of characteristic absorptions indicate the formation of two new products. Based on previously reported data10 and on the basis of results of our present studies on the reaction of the allyl radical generated by HVFP of 1,5-hexadiene with molecular oxygen, one of them was readily identified as the allyldioxyl radical (10) (Fig. 1). Another new product with an intense band located at 1191.1 cm−1 and less intense at 1295.5 and 686.3 cm−1, respectively, was identified as 5. Conceivable alternative structures resulting from bonding to the O-atom do not represent energy minima (ESI,† Fig. S1).
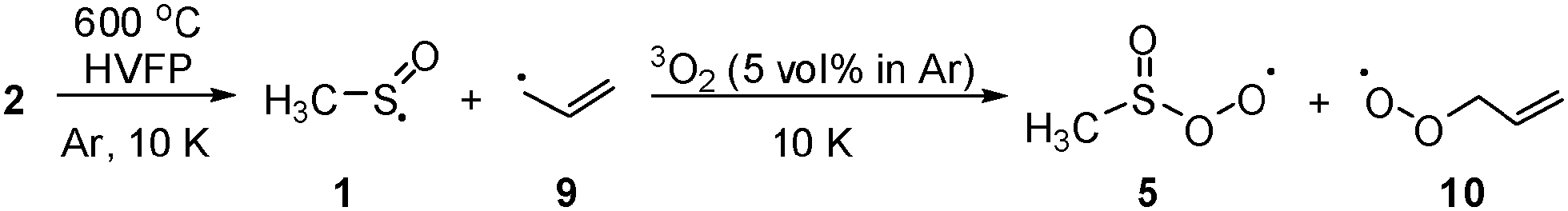 | ||
| Scheme 4 Generation of the methylsulfinyl radical (1) by HVFP of precursor 3 and its reaction with oxygen in solid argon at 10 K. | ||
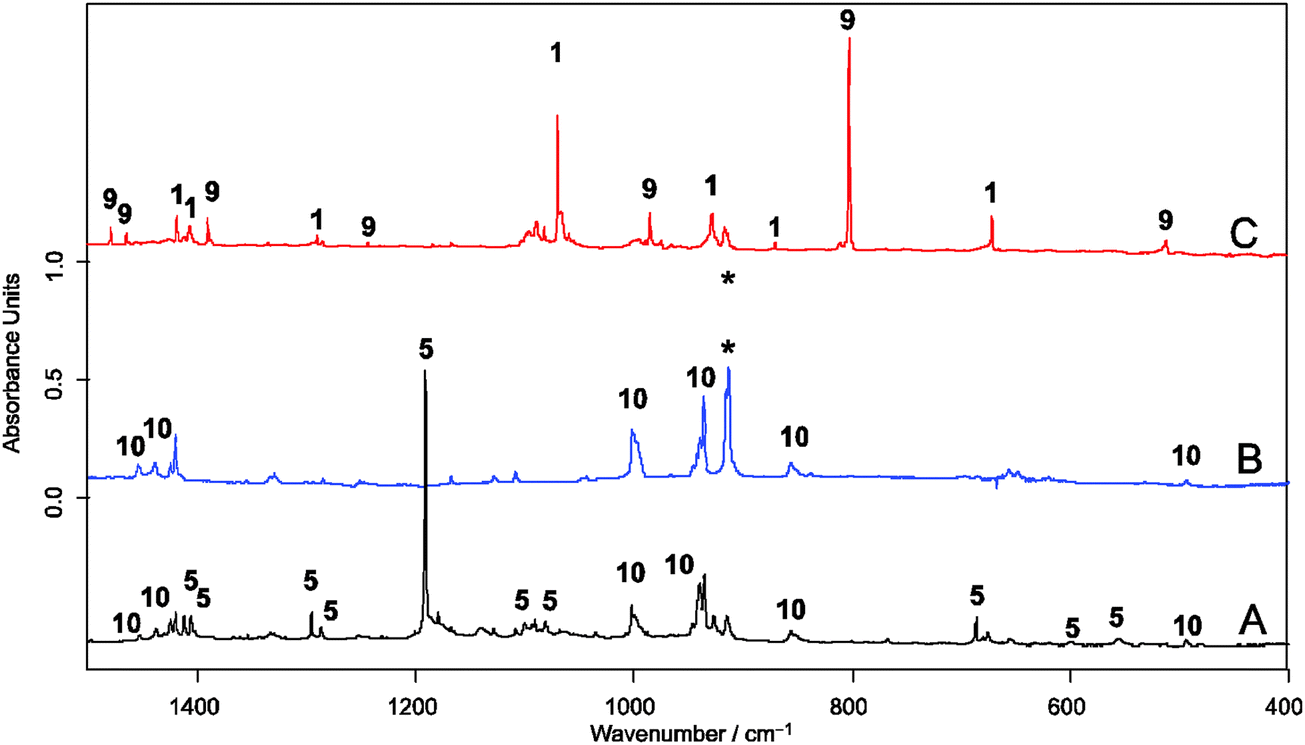 | ||
| Fig. 1 FTIR spectra of pyrolysis of precursor 2 (A) and 1,5-hexadiene (*) (B) isolated in an Ar matrix containing 5.0 vol% 3O2; pyrolysis products of 2 isolated in an Ar matrix in the absence of 3O2 (C). | ||
The UV/Vis spectrum of the matrix material showed a broad absorption maximum at λmax = 394 nm (Fig. 2, full line). Based on this observation, photolyses (Hg high pressure lamp, monochromator, Δλ = 10 nm) were carried out with light at λ = 366 nm or alternatively at λ = 405 nm. In both cases, disappearance of the initial IR- and UV/Vis absorption bands attributed to 5 was observed, whereas those of 10 remained unchanged. The products formed thereby were identified as SO3 and 6. The presence of small amounts of SO3 is evident from its characteristic IR bands at 1391/1383 cm−1, 527/521 cm−1, and 489 cm−1.11 The fate of the methyl radical as the co-product of the photodissociation reaction remains unclear. Neither its IR bands nor those of the methylperoxyl radical12 as the expected reaction product with excess 3O2 could be found, most probably because their specific intensities are much lower than that of SO3. The structure of the second product was deduced on the basis of its UV/Vis spectrum revealing a very strong absorption band in the range of 510–350 nm with a distinct vibrational fine structure along with a much weaker broad structureless absorption between ca. 700 nm and 530 nm (Fig. 2, dashed line). Similar bands were reported in a laser flash photolysis experiment for the initial photodissociation product of bis(methylsulfonyl)peroxide and assigned to 6.13 Moreover, similar bands were observed for the product obtained from the same precursor after HVFP with subsequent isolation in noble gas matrices.14
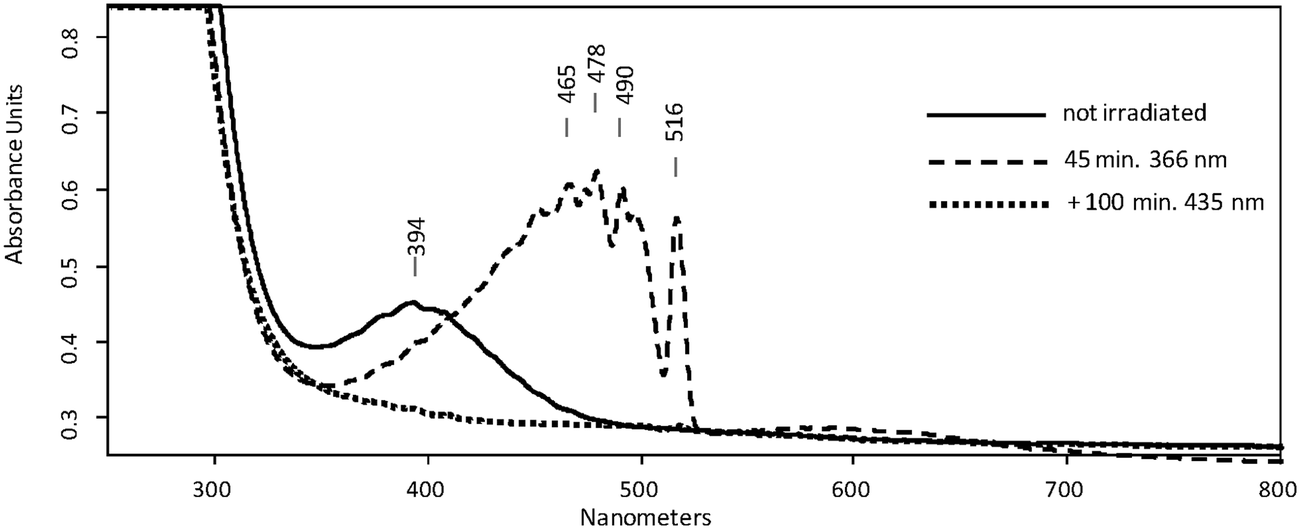 | ||
| Fig. 2 UV/Vis spectra of pyrolysis of precursor 2 isolated in an Ar matrix containing 5.0 vol% 3O2. | ||
In good accordance with the experimental UV/Vis spectra, TD-UB3LYP computations of the excitation energies (ESI,† Table S3) provide two weak absorption bands at 388 and 366 nm for 5 and a very intense band at 476 nm for 6, whereas the photochemical final product 7 should have no absorption above 315 nm. For the unobserved isomer 8 the computations predict absorptions of medium intensity at 375 and 361 nm that are close to those of 5. It might well be that 8 forms upon irradiation, but it is photolabile and therefore its concentration remains below the detection limit.
The most intense IR bands attributed to 6 were observed at 757, 1334, and 1441 cm−1 (Fig. 3, negative bands). They coincide well with those reported for argon matrix isolated 6.14 Comparison of the experimental IR bands with computed IR spectra of 6 was not helpful in supporting the structural assignment. Especially the theoretically predicted strong band for the symmetric SO3 stretching vibrations (a1) has no counterpart in the experimental spectrum. Due to the special electronic situation of C3v symmetric 6 with a degenerate singly occupied highest molecular orbital that is subject to Jahn–Teller distortion to Cs symmetry, one may expect strong vibronic coupling of the energy levels and anharmonic potentials. The electronic situation of 6 is similar to that of the highly symmetric NO3 radical for which it was shown that standard DFT and ab initio methods completely fail in predicting the vibrational frequencies due to vibronic coupling and highly anharmonic potentials.15
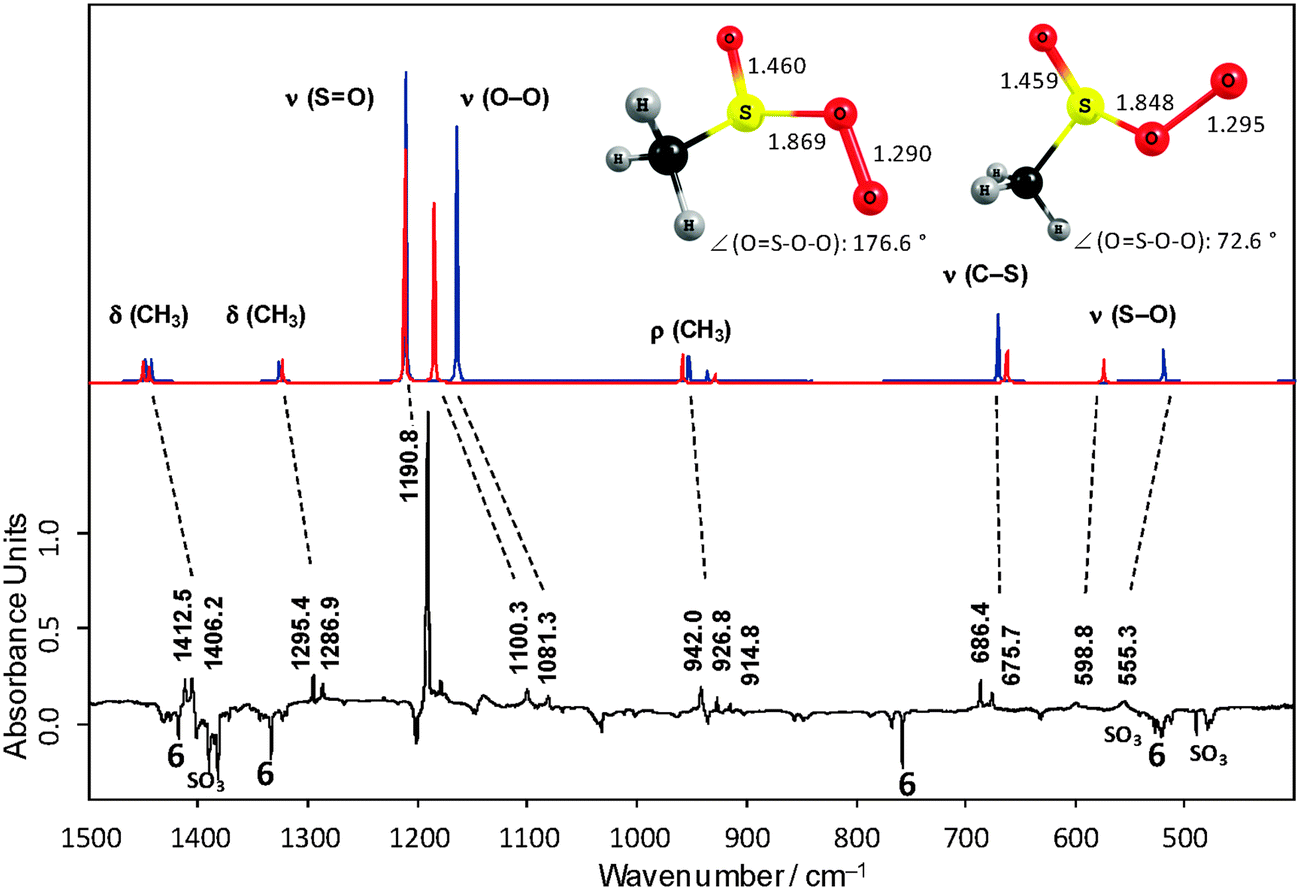 | ||
| Fig. 3 Comparison of the computed IR spectra of two conformers of 5 (UB3LYP/6-311+G(3df,3pd) unscaled, above) and the experimental difference spectrum of the 366 nm photolysis of the 3O2 adduct 5 (below); positive bands vanish, negative bands appear upon irradiation. | ||
Prolonged irradiation of the matrix with light at λ = 435 nm showed the disappearance of 6, and no absorption maximum could be found in the UV/Vis spectrum above 300 nm. The IR spectra of the new product can be assigned to C-centered radical 7. The absorption band located at 3569 cm−1 unambiguously proves the formation of the –S(O)2OH group. This position is very close to OH-stretching vibration of argon matrix isolated H2SO4 at 3567 cm−1 and CH3SO3H at 3579 cm−1, respectively.16 In addition, absorptions at 1403, 1391, 1383, 1189, 1184, 842, 827, 784 cm−1, partially split by matrix effects, can also be attributed to 7 (ESI,† Fig. S3). Upon labelling two of the three oxygen atoms of 7 by 18O, two distinct OH stretching bands at 3570.0 and 3559.0 cm−1 with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were observed (ESI,† Fig. S4) as expected for the postulated structure. Apparently, the thermodynamically most stable isomeric radical 8 does not form in the matrix under photolysis. Neither 8 nor the expected decomposition products such as the methoxy radical and SO2 could be detected.
:2 were observed (ESI,† Fig. S4) as expected for the postulated structure. Apparently, the thermodynamically most stable isomeric radical 8 does not form in the matrix under photolysis. Neither 8 nor the expected decomposition products such as the methoxy radical and SO2 could be detected.
Structure 5 is expected to exist as a mixture of rotamers by rotation around the S–O single bond. The results of DFT and ab initio computations show that they differ by 0.5 kcal mol−1 (see data for 5 in ESI†). The computed IR spectrum of equal amounts of two representative rotamers fits well to the experimental spectrum (Fig. 3). The most intense band observed at 1191 cm−1 is attributed to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond stretch. Apparently, within the applied experimental spectral resolution the positions of the ν(SO) absorption bands are identical for both rotamers.
O double bond stretch. Apparently, within the applied experimental spectral resolution the positions of the ν(SO) absorption bands are identical for both rotamers.
The formation of 5 as the initial product in the reaction of 1 with 3O2 is strongly supported by additional isotopic labelling experiments. In particular, the observed spectral shifts in the IR spectrum of the 18O-labelled product were key for the spectral assignments (Fig. 4; in ESI,† Tables S1 and S2).
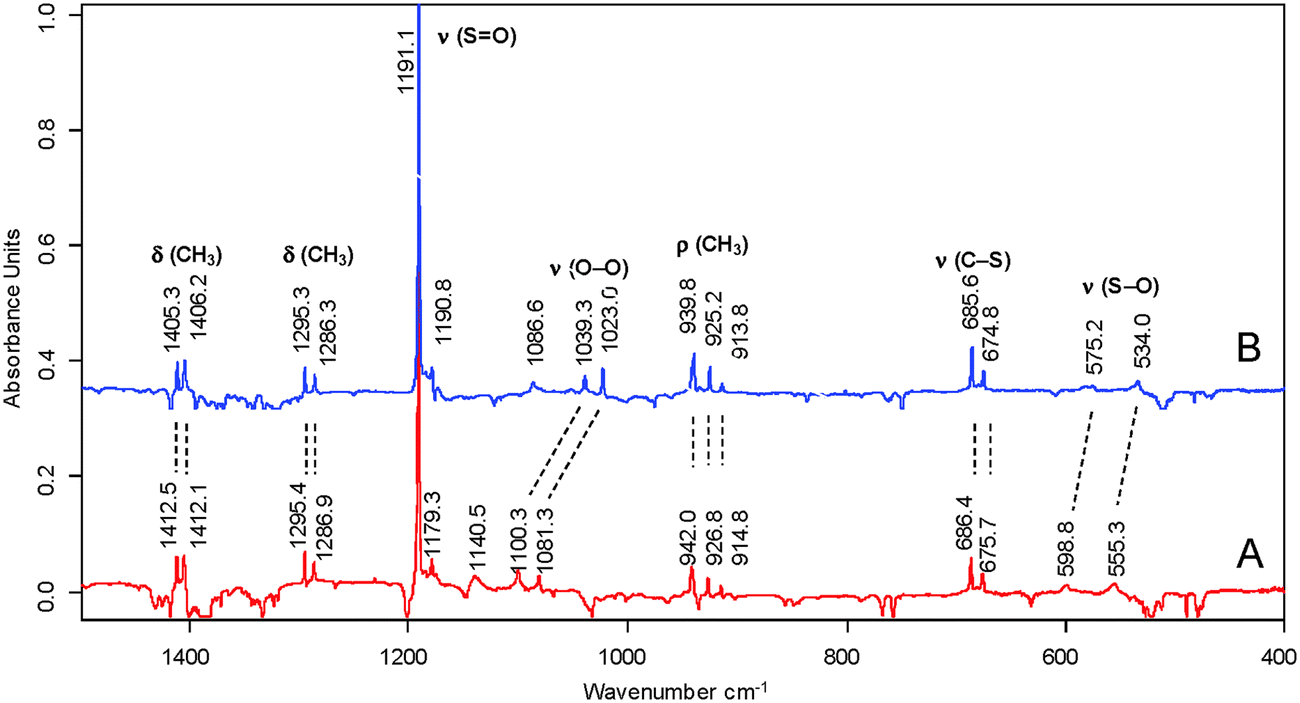 | ||
| Fig. 4 Isotopical band shifts of the [18O2]-methylsulfinyldioxy radical ([18O2]-5); radicals 5 were obtained by reaction of methylsulfinyl radical (1) with triplet 16O2 (A) and triplet 18O2 (B). | ||
The most intense IR band of 5 displayed only a very small isotopic band shift (+0.2 cm−1) when triplet 18O2 was used instead of triplet 16O2. This IR band could be attributed unambiguously to a pure SO stretching vibration. Furthermore, the labelling experiment provides evidence that a pair of weak bands at 1100 and 1082 cm−1 has to be assigned to the O–O stretching vibrations of two rotamers of 5. The experimental band shifts of −61.0 and −58.0 correspond well to the isotopic shifts (ESI,† Tables S1 and S2). Moreover, the expected band shifts were also observed for the S–O stretching vibration (600 cm−1 and 555 cm−1versus 575 cm−1 and 534 cm−1, respectively). The observed IR band shifts after deuteration of the methyl group (ESI,† Fig. S5) also agree very well with these band assignments. Whereas, in accordance with DFT computation (ESI,† Fig. S6), the positions of the ν(SO) and the ν(O–O) remain almost unchanged, and large bathochromic shifts were observed for the δ(CH3) and ρ(CH3) vibrations.
The present study shows that atmospherically highly relevant radical 1 generated in the gas phase by high-vacuum flash pyrolysis reacts spontaneously with molecular oxygen in a solid argon matrix under cryogenic conditions. Obviously, the formation of the oxygen adduct is a nearly barrierless, exothermic process. The structure of initially formed peroxyl radical 5 was proven by comparison of computed with experimental IR and UV spectra as well as through extensive isotopic labelling experiments. Upon irradiation with near UV light, adduct 5 undergoes photochemical dissociation into SO3 and a methyl radical or isomerization to 6. The latter is also not stable against irradiation with visible light and eventually transforms to 7. These results underscore earlier postulates regarding the existence of a reactive oxygen adduct of the methylsulfinyl radical. Furthermore, our results demonstrate the importance of photochemical transformations in atmospheric oxidation processes of volatile organic sulfur compounds. These photochemical pathways have not yet been considered in the discussions related to the atmospheric sulfur cycle.
This work was supported by the DAAD Partnership of the University of Lodz and the Justus-Liebig University. G. M. thanks National Science Center (Poland) for financial support (Project Meastro-3; Dec-2012/06/A/ST5/00219).
Notes and references
- J. E. Lovelock, J. Maggs and R. A. Rasmussen, Nature, 1972, 237, 452–453 CrossRef CAS PubMed.
- T. S. Bates, B. K. Lamb, A. Guenther, J. Dignon and R. E. Stoiber, J. Atmos. Chem., 1992, 14, 315–337 CrossRef CAS.
- (a) R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Nature, 1987, 326, 655–661 CrossRef CAS PubMed; (b) G. P. Ayers and J. M. Cainey, Environ. Chem., 2007, 4, 366–374 CrossRef CAS; (c) I. Faloona, Atmos. Environ., 2009, 43, 2841–2854 CrossRef CAS PubMed.
- (a) G. S. Tyndall and A. R. Ravishankara, Int. J. Chem. Kinet., 1991, 23, 483–527 CrossRef CAS PubMed; (b) J. Barnes, J. Hjorth and N. Mihalopoulos, Chem. Rev., 2006, 106, 940–975 CrossRef PubMed.
- H. P. Reisenauer, J. Romański, G. Mlostoń and P. R. Schreiner, Chem. Commun., 2013, 49, 9467–9469 RSC.
- H. P. Reisenauer, P. R. Schreiner, J. Romański and G. Mlostoń, J. Phys. Chem. A, 2015, 119, 2211–2216 CrossRef CAS PubMed.
- (a) G. S. Tyndall and A. R. Ravishankara, J. Phys. Chem., 1989, 93, 2426–2435 CrossRef CAS; (b) F. Yin, D. Grosjean, R. C. Flagan and J. H. Seinfeld, J. Atmos. Chem., 1990, 11, 365–399 CrossRef CAS.
- S. B. Barone, A. A. Turnipseed and A. R. Ravishankara, Faraday Discuss., 1995, 100, 39–54 RSC.
- Z. Salta, A. M. Kosmas and A. Lesar, Comput. Theor. Chem., 2012, 1001, 67–76 CrossRef CAS PubMed.
- E. G. Baskir and O. M. Nefedov, Russ. Chem. Bull., 1996, 45, 99–106 CrossRef.
- (a) V. E. Bondybey and J. H. English, J. Mol. Spectrosc., 1985, 109, 221–228 CrossRef CAS; (b) A. Givan, A. Loewenschuss, C. J. Nielsen and M. Rozenberg, J. Mol. Struct., 2007, 830, 21–34 CrossRef CAS PubMed.
- (a) P. Ase, W. Bock and A. Snelson, J. Phys. Chem., 1986, 90, 2099–2109 CrossRef CAS; (b) S. Nandi, S. J. Blanksby, X. Zhang, M. R. Nimlos, D. C. Dayton and G. B. Ellison, J. Phys. Chem. A, 2002, 106, 7547–7556 CrossRef CAS.
- H.-G. Korth, A. G. Neville and J. Lysztyk, J. Phys. Chem., 1990, 94, 8835–8839 CrossRef CAS.
- X. Zeng, H. Beckers, J. S. Francisco, H. Willner, to be submitted (private communication).
- (a) J. F. Stanton, Mol. Phys., 2009, 107, 1059–1075 CrossRef CAS PubMed; (b) J. F. Stanton, J. Gauss and R. J. Bartlett, J. Phys. Chem., 1991, 94, 4084–4087 CrossRef CAS PubMed; (c) J. F. Stanton, J. Chem. Phys., 2007, 126, 134309 CrossRef PubMed.
- A. Givan, A. Loewenschuss and C. J. Nielsen, J. Mol. Struct., 2005, 748, 77–90 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table, experimental, additional IR spectra and computational details. See DOI: 10.1039/c5cc02168e |
| This journal is © The Royal Society of Chemistry 2015 |