
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic graphene oxide stabilisation of hexagonal BN and MoS2 sheets†
M. Haniff
Wahid
ab,
Xianjue
Chen
*a,
Christopher T.
Gibson
a and
Colin L.
Raston
*a
aCentre for NanoScale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Bedford Park, SA 5042, Australia. E-mail: colin.raston@finders.edu.au; Tel: +61 88201 7958
bDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
First published on 17th June 2015
Abstract
A simple and scalable method has been developed for directly forming water-dispersible van der Waals solids involving mixing aqueous solution of graphene oxide (GO) with hexagonal boron nitride (BN) or molybdenum disulphide (MoS2) in N-methylpyrrolidone. The GO acts as an amphiphile in stabilising the colloidal solutions of the heterolaminar material in water.
The breakthrough in unveiling the exceptional properties of graphene has triggered extensive research efforts in two-dimensional (2D) materials, including inorganic layered analogues such as hexagonal boron nitride (BN), and molybdenum disulfide (MoS2), and other transition metal dichalcogenides, and layered oxides.1 In addition to investigating the behaviour of monolayers, there is a rapidly emerging focus on constructing or reassembling isolated layers of such material into designer multi-layer heterostructures that are bound together primarily by van der Waals forces.2 Such cohesion forces essentially preserve the distinct electronic properties of individual layers of the material,3 enabling tunable properties of the heterostructures, with potential for functioning as high performance electronic switches and optoelectronic devices that are still challenging to access for graphene due to the absence of an intrinsic energy bandgap.4
Advancing the practical applications of the above heterolaminar van der Waals materials requires the development of scalable syntheses. In comparison to the well-developed strategies for fabricating graphene and quasi-graphene forms (graphene oxide or reduced graphene oxide), the possibility of making multi-layer van der Waals solids has only recently been achieved experimentally.5,6 Significant efforts have been made towards growing graphene, monolayer BN and MoS2 epitaxially on top of each other with controllable quality, although finding the right conditions for growing continuous layers is challenging.7–9 Another approach is layer-by-layer deposition from 2D material suspensions via Langmuir–Blodgett techniques.10,11 It is also possible to mix preformed colloidal suspensions of different 2D materials to generate layered flocculates.12 This solution-based self-organising method is versatile and scalable, showing promise for producing van der Waals solids as ultrathin dielectrics,10 selectively permeable membranes,13 and composite materials.14 However, there are some limitations on the present approaches which involve the use of specific organic solvents that have similar surface tension to that of the stabilised 2D materials.15,16 Organic solvents can only ensure temporary stability and finite concentration of the 2D material suspensions,17 and thus can restrict further processing of such materials. It is also difficult to produce stabilised inorganic lamellar materials, including BN and MoS2, in aqueous media in the absence of additional surfactants due to their hydrophobic property.18,19 Indeed access to water-processable quasi-graphene forms such as graphene oxide and reduced graphene oxide has dramatically advanced the potential of graphene-based materials into much broader areas.20,21 Hence, there is significant scope for developing novel and versatile solution-based protocols to prepare water-dispersible heterolaminar van der Waals materials.
In this work we develop the use of graphene oxide (GO) sheets as both the composite material and the surfactant for supporting and stabilising BN and MoS2 sheets in water. GO is amphiphilic due to the presence of carboxyl and hydroxyl groups at the edges and hydrophobic unoxidized polyaromatic islands within the basal plane.22,23 This amphiphilic property has been used for effectively stabilising pristine graphite flakes and carbon nanotubes,23 creating highly stable Pickering emulsions,23,24 and constructing three-dimensional networks of GO hollow spheres.25 This suggests that GO is capable of non-covalently assembling with BN or MoS2 sheets via van der Waals interactions and/or hydrophobic effects, at the same time stabilising the heterolaminar materials in water, and this is the focus of the work reported herein.
The overall procedure for preparing the heterolaminar materials is summarised in Fig. 1. GO was prepared from high purity graphite flakes powder (99.9%, SP-1, Bay Carbon) using the modified Hummer's method.‡26,27 Colloidal dispersions (10 mL) of BN (1 mg mL−1) and MoS2 (1 mg mL−1) in N-methylpyrrolidone (NMP) were prepared using probe sonication for 1 hour. Each dispersion was then diluted to 0.2 mg mL−1 (0.5 mL) and directly added drop-wise into 0.5 mL of colloidal suspension of GO (0.5 mg mL−1), followed by mild sonication for 30 seconds. The stability of the suspensions were investigated under acidic (pH 1), neutral and basic (pH 12) conditions. We selected a relatively low starting concentration for BN and MoS2 dispersions, because higher concentrations were observed to induce instability of the mixed solutions, presumably due to an increase in irreversible loading of BN or MoS2 sheets onto GO sheets. The resulting mixed dispersions were centrifuged (860 × g, 15 min) to remove any large flakes of the starting laminar materials, with the supernatants then separated, affording the colloidal dispersions of GO–BN and GO–MoS2. Centrifugation of samples prepared under acidic condition showed complete precipitation, and thus loss of stability of the material (ESI-S1†). NMP was the dispersing medium of choice for the exfoliation and stabilization due to the insolubility of BN and MoS2 in water, but not in NMP, as well as the infinite miscibility of NMP with water. Despite the ability to directly exfoliate BN and MoS2 in the presence of GO, pre-sonication of BN and MoS2 in NMP effectively avoids any risk of damaging the structure of GO. The NMP in the resulting solutions can be further removed by centrifugal-washing the samples at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370 × g for 1 hour, and redispersing the sediments into water, affording stable dispersions, Fig. 1. The colour difference between the solutions after mixing and after re-dispersing arises from the higher stability of the GO devoid of BN or MoS2, whereas the heterolaminar materials are more readily separated centrifugally.
370 × g for 1 hour, and redispersing the sediments into water, affording stable dispersions, Fig. 1. The colour difference between the solutions after mixing and after re-dispersing arises from the higher stability of the GO devoid of BN or MoS2, whereas the heterolaminar materials are more readily separated centrifugally.
 | ||
| Fig. 1 Schematic illustration of the sample preparation and corresponding photographs of the resulting dispersions. | ||
Control experiments involving mixing NMP solutions of BN and MoS2 with water showed no evidence for forming stable dispersions, clearly establishing that the formation of any dispersions in water requires the presence of GO. This supports our hypothesis that GO acts as an amphiphilic surfactant, in binding to BN and MoS2 through the hydrophobic islands in lowering their interfacial energy, with hydrophilic groups on the outer surface of the GO stabilising the colloidal dispersion of the heterolaminar material in water. Pertaining to the effect of pH, under acidic conditions there is a loss in stability of the GO (ESI-S1†) whereas stable dispersions formed under neutral and basic (pH 12) conditions. The electrostatic stability of the re-dispersed solutions of GO–BN and GO–MoS2 at neutral pH was confirmed by zeta potentials, at −48.3 mV, −47.7 mV and −50.3 mV, respectively, for GO solution, and re-dispersed solutions of GO–BN and GO–MoS2.
Transmission electron microscopy (TEM) was used to investigate the state of the re-dispersed composite materials in water, by dropping dispersions onto a holey carbon coated copper grid and drying under ambient conditions. TEM images for pristine GO, exfoliated BN and MoS2 are shown in Fig. 2a–d, respectively. Graphene oxide sheets (∼200 nm to 10 μm in lateral dimensions) are evident in Fig. 2a and 3a, with the corresponding electron diffraction (inset) revealing a typical pattern for monolayers. The pattern rings of electron diffraction in the inset of Fig. 2b are consistent with randomly stacked layers of GO.28 TEM images and corresponding electron diffraction patterns from selected areas in Fig. 2c and d, for as prepared separate NMP solutions of BN and MoS2, confirm the presence of exfoliated BN and MoS2 sheets. The exfoliated sheets range in size from ∼200 nm to 1 μm for both materials. While BN gives a similar shape and size due to the smaller cross section of the starting material (∼1 μm), MoS2 (∼2 μm) shows an apparent fragmentation, presumably arising from high energy cavitation processes associated with the sonication and less strong in-plane stiffness compared with BN sheets.29Fig. 2e and g show the composites of GO–BN and GO–MoS2 in the re-dispersed solutions, respectively, with the size of the BN and MoS2 consistent with that of the as-exfoliated sheets in NMP. Similar images were obtained for samples prepared under basic conditions (ESI-S2†). The electron diffraction pattern in Fig. 2f is similar to that of the stacking monolayers in Fig. 2b. Even though it is difficult to determine the overlapping diffraction spots of BN in this pattern, as BN and graphene have very similar crystal structures with a lattice constant difference of ∼2%, the difference in contrast, size and shape of BN and GO sheets provides a means to recognise BN. In the case of GO–MoS2, the electron diffraction (Fig. 2h) shows a typical pattern for MoS2, along with the GO ring pattern. The TEM analysis also shows that BN and MoS2 always accompany GO, which is in accordance with the BN and MoS2 sheets being supported on the surface of GO in the re-dispersed solutions.
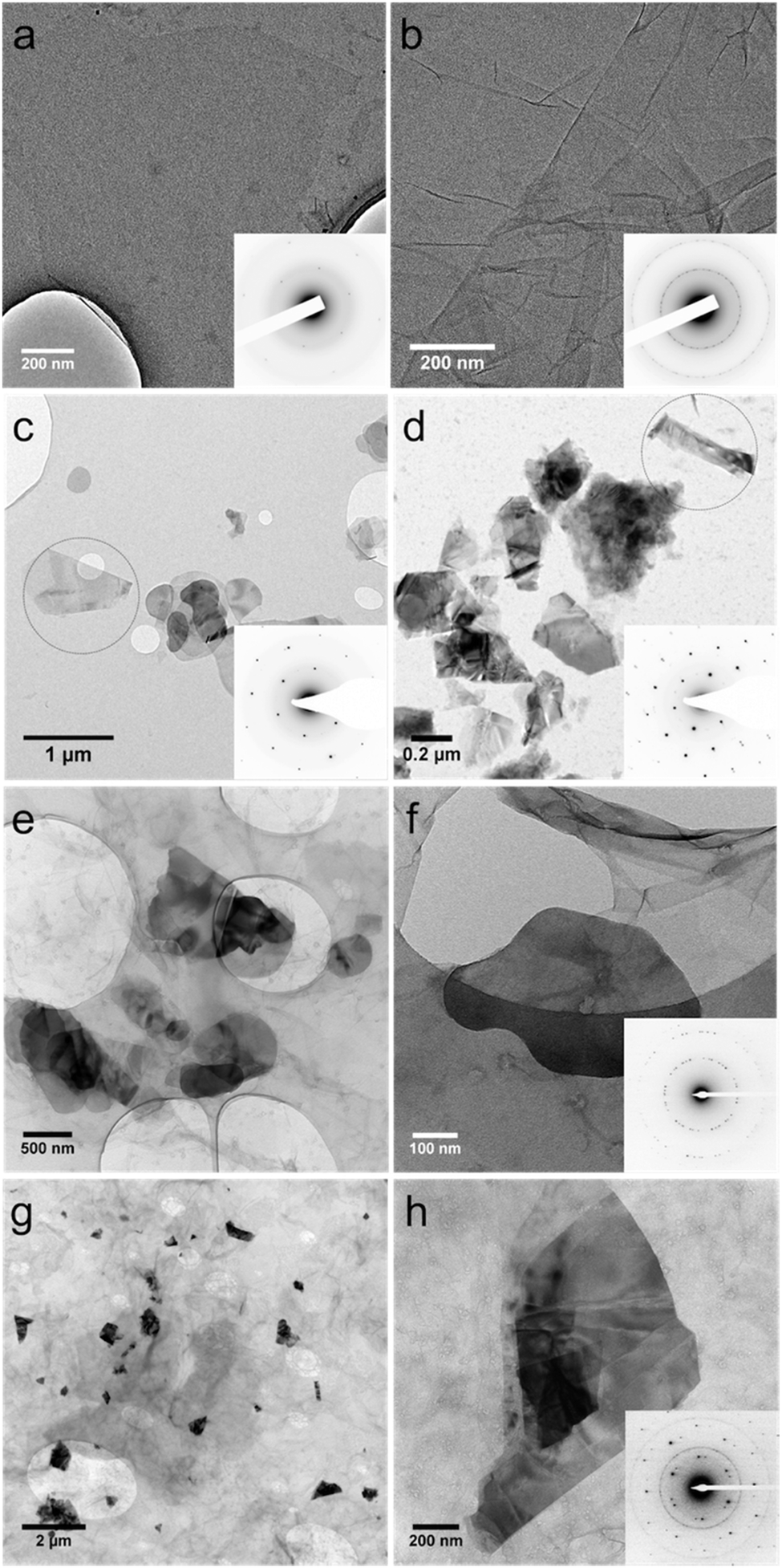 | ||
| Fig. 2 TEM images of (a, b) GO, (c) exfoliated BN and (d) MoS2 sheets, (e, f) GO–BN and (g, h) GO–MoS2. Electron diffraction patterns for all samples are as shown in the inset. | ||
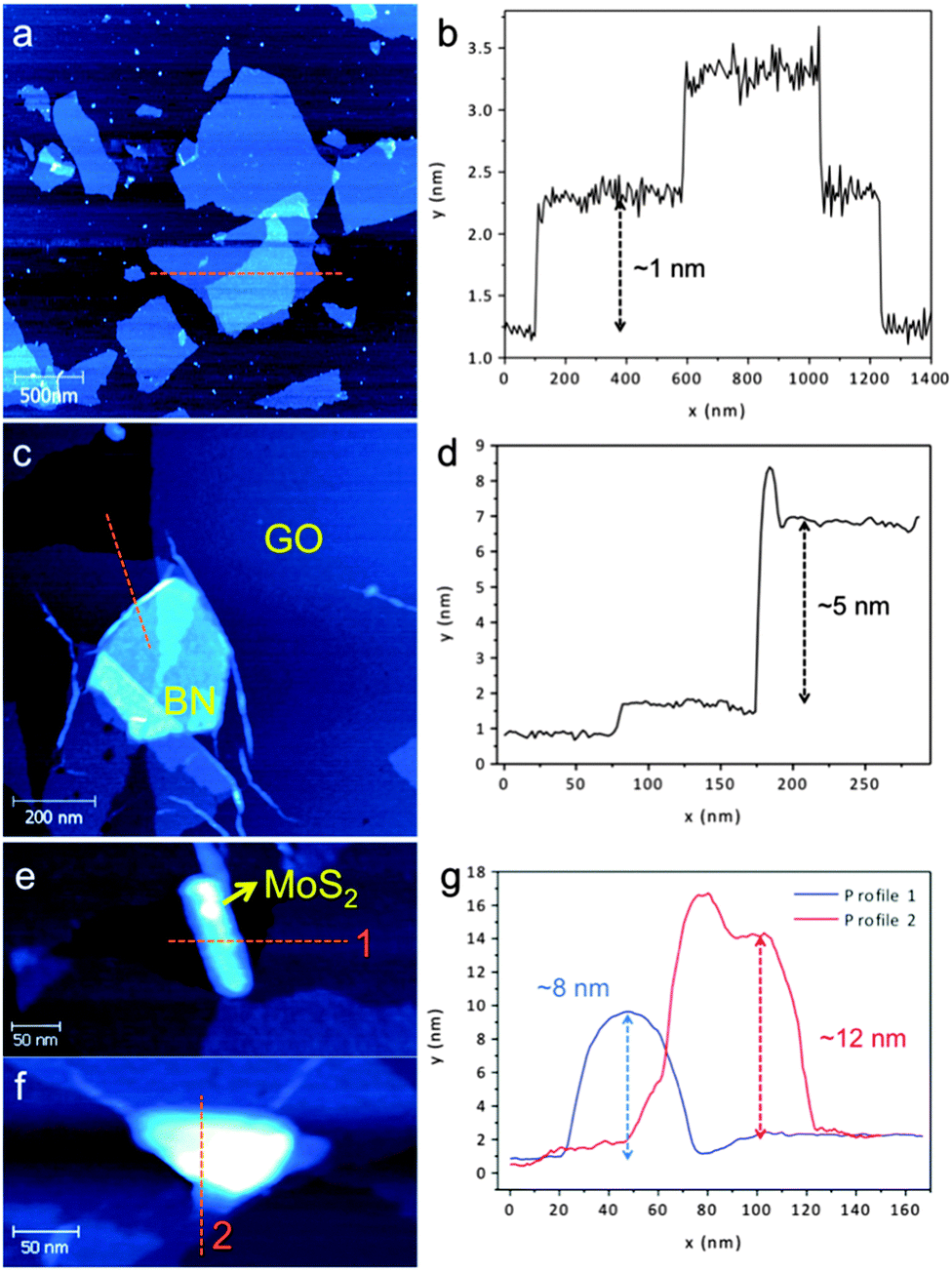 | ||
| Fig. 3 AFM images and height profiles of the indicated area in (a, b) GO, (c, d) GO–BN, and (e, f, g) MoS2 samples. | ||
Atomic force microscopy (AFM) was undertaken to further investigate the morphology of the heterolaminar materials. The pristine GO is monolayered having a measured thickness of ∼1 nm,21Fig. 3a and b. The structures of heterolaminar materials are consistent with TEM analysis, with BN and MoS2 supported on GO evident in Fig. 3c, e and f. The height profile measurement in Fig. 3d along the orange line in Fig. 3c reveals a thickness of ∼5 nm for BN. The height profiles for the GO–MoS2 show that the sheets of MoS2 are ∼8–12 nm thick, Fig. 3g. The difference in thickness is related to the different interplanar distances between BN (∼0.33 nm) and MoS2 (∼0.61 nm), as well as the higher surface energy of MoS2 (>250 mJ m−2) compared to that of BN (∼110 mJ m−2),30,31 requiring higher energy input for exfoliation.
Raman spectra were recorded for pristine GO, BN and MoS2, and for the re-dispersed heterolaminar materials respectively (Fig. 4). Typical spectra for GO displayed a D and G band at ∼1370 cm−1 and ∼1570 cm−1 respectively (Fig. 4a).32 For BN (Fig. 4b) and MoS2 (Fig. 4c), strong fluorescence background was observed, possibly due to the remaining NMP which was used as the dispersant. BN exhibits a characteristic peak at ∼1360 cm−1 that is due to the E2g phonon mode which is similar to the G peak in graphitic materials, as shown in the zoomed-in image in Fig. 4(i).33 MoS2 has characteristic peaks at ∼380 cm−1 and ∼400 cm−1 which are attributed to the E12g and A1g modes, respectively, Fig. 4(ii).34 Strong D and G bands for GO are evident for both GO–BN and GO–MoS2. The characteristic peak for BN cannot be seen due to overlap with the strong D band for GO, whereas for MoS2, both characteristic peaks are discernible, Fig. 4(iii).
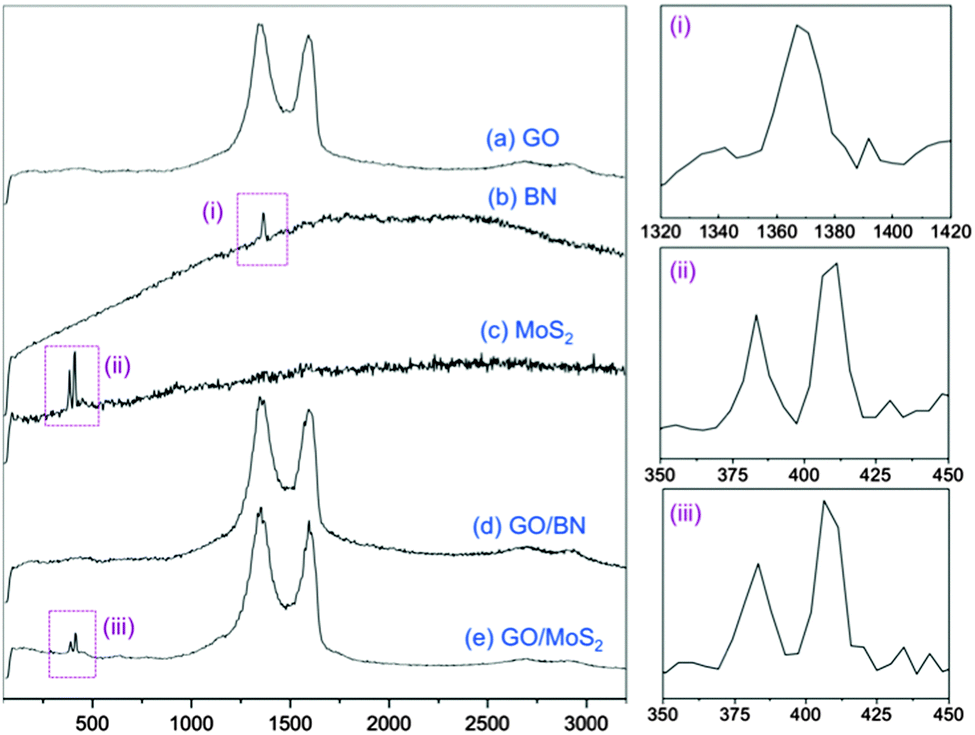 | ||
| Fig. 4 Raman spectra of pristine (a) GO, (b) BN, (c) MoS2 and composite material of (d) GO–BN and (e) GO–MoS2, respectively. Zoomed-in images of the peaks labelled as (i), (ii) and (iii) are shown on the right. | ||
In conclusion, we have developed a facile method for preparing heterolaminar van der Waals composites based on graphene oxide and BN or MoS2 in water. This involves a simple sonication of BN and MoS2 in NMP as a dispersing medium, for then adding to preformed GO in water, with the subsequent processing leading to aqueous solutions devoid of organic solvent. Importantly the method can be readily scaled into gram quantities of the material. Moreover, this work extends the utility of GO in stabilising 2D layered materials in water and consequently it opens up new facile processing adventures for 2D layered materials in general, particularly in water.
We gratefully acknowledge support of this work by the Australian Research Council and the Government of South Australia. Valuable discussions with Dr Ramiz A. Boulos on this work is greatly appreciated. M.H. Wahid would like to thank the Malaysian Government and Universiti Putra Malaysia for the PhD research funding. TEM studies were carried out using facilities in Adelaide Microscopy at The University of Adelaide. AFM and Raman analyses were carried out at Flinders University, supported by the Australian Microscopy and Microanalysis Research Facility (AMMRF). The authors also would like to thank Dr Sheng Dai for the assistance in zeta potential analysis.
Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419 CrossRef CAS PubMed.
- B. Radisavljevic and A. Kis, Nat. Mater., 2013, 12, 815 CrossRef CAS PubMed.
- G. S. Duesberg, Nat. Mater., 2014, 13, 1075 CrossRef CAS PubMed.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim, K. L. Shepard and J. Hone, Nat. Nanotechnol., 2010, 5, 722 CrossRef CAS PubMed.
- L. A. Ponomarenko, A. K. Geim, A. A. Zhukov, R. Jalil, S. V. Morozov, K. S. Novoselov, I. V. Grigorieva, E. H. Hill, V. V. Cheianov, V. I. Fal’co, K. Watanabe, T. Taniguchi and R. V. Gorbachev, Nat. Phys., 2011, 7, 958 CrossRef CAS PubMed.
- T. Tanaka, A. Ito, A. Tajiima, E. Rokuta and C. Oshima, Surf. Rev. Lett., 2003, 10, 721 CrossRef CAS.
- Z. Liu, L. Song, S. Zhao, J. Huang, L. Ma, J. Zhang, J. Lou and P. K. Ajayan, Nano Lett., 2011, 11, 2032 CrossRef CAS PubMed.
- Y. Shi, W. Zhou, A.-Y. Lu, W. Fang, Y.-H. Lee, A. L. Hsu, S. M. Kim, K. K. Kim, H. Y. Yang, L.-J. Li, J.-C. Idrobo and J. Kong, Nano Lett., 2012, 12, 2784 CrossRef CAS PubMed.
- M. Osada and T. Sasaki, Adv. Mater., 2012, 24, 210 CrossRef CAS PubMed.
- K. Ariga, Q. Ji, J. P. Hill, Y. Bando and M. Aono, NPG Asia Mater., 2012, 4, e17 CrossRef PubMed.
- G. Gao, W. Gao, E. Cannuccia, J. Taha-Tijerina, L. Balicas, A. Mathkar, T. N. Narayanan, Z. Liu, B. K. Gupta, J. Peng, Y. Yin, A. Rubio and P. K. Ajayan, Nano Lett., 2012, 12, 3518 CrossRef CAS PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442 CrossRef CAS PubMed.
- R. J. Young, I. A. Kinloch, L. Gong and K. S. Novoselov, Compos. Sci. Technol., 2012, 72, 1459 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGoVern, B. Holland, M. Byrne, Y. K. Gun’ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O’Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Mariarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568 CrossRef CAS PubMed.
- W. W. Liu and J. N. Wang, Chem. Commun., 2011, 47, 6888 RSC.
- X. Chen, R. A. Boulos, P. K. Eggers and C. L. Raston, Chem. Commun., 2012, 48, 11407 RSC.
- X. Chen, W. Zang, K. Vimalanathan, K. S. Iyer and C. L. Raston, Chem. Commun., 2013, 49, 1160 RSC.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180 CrossRef CAS PubMed.
- S. C. Thickett and P. B. Zetterlund, ACS Macro Lett., 2013, 2, 630 CrossRef CAS.
- X. Chen, P. K. Eggers, A. D. Slattery, S. G. Ogden and C. L. Raston, J. Colloid Interface Sci., 2014, 430, 174 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- J.-K. Lee, S. Lee, Y.-I. Kim, J.-G. Kim, B.-K. Min, K.-I. Lee, Y. Park and P. John, Sci. Rep., 2014, 4, 5682 CAS.
- C. Ataca, H. Şahin, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2011, 115, 3934 CAS.
- K. Weiss and J. M. Philips, Phys. Rev. B: Solid State, 1976, 14, 5392 CrossRef CAS.
- K. M. Knowles and S. Turan, Cryst. Res. Technol., 2000, 35, 751 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- R. V. Gorbachev, I. Riaz, R. R. Nair, R. Jalil, L. Britnell, B. D. Belle, E. W. Hill, K. S. Novoselov, K. Watanabe, T. Taniguchi, A. K. Geim and P. Blake, Small, 2011, 7, 465 CrossRef CAS PubMed.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc02066b |
| ‡ Methods summary: GO used in this study was prepared from natural graphite flakes powder (99.9%, SP-1, Bay Carbon) using the modified Hummer's method.26,27 Probe sonication was carried out using a Vibra-Cell™ VCX130 sonicator at 60% amplitude for all samples. Centrifugations were performed using the Dynamica Velocity 14R. Zeta potential analysis was performed using a Malvern Zetasizer Nano series. Measurements for each sample were recorded in triplicate and 20 data acquisitions were recorded in each measurement. All measurements were recorded at 25 °C in Malvern disposable clear Folded Capillary Cells. TEM samples were prepared by depositing a drop of the suspension onto a holey carbon coated copper grid (#2450-AB, SPI Supplies) and dried under ambient conditions. TEM analysis was carried out using a Philips CM200 instrument operating at 120–200 kV. Image J software was used for processing all the TEM images. Raman spectra were acquired using a Witec alpha300R Raman microscope with excitation laser wavelength of 532 nm (≤5 mW), at room temperature. The spectra were recorded with an ×40 objective (Numerical Aperture 0.60) for each sample with typical integrations times between 10 to 20 seconds with 3 accumulations per spectrum. Atomic force microscopy (AFM) analysis in tapping mode was performed with a Bruker multimode AFM and a NanoScope V under ambient conditions. AFM probes used were Mikromasch HQ:NSC15 Si probes with a nominal spring constant of 40 N m−1 and a nominal tip diameter of 16 nm. The AFM scanner was calibrated in the x, y and z axes using Si calibration grids (Bruker model numbers PG: 1 μm pitch, 110 nm depth, and VGRP: 10 μm pitch, 180 nm depth). Samples were deposited on mica substrates and dried in air prior to analysis. |
| This journal is © The Royal Society of Chemistry 2015 |