
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oscillatory three-phase flow reactor for studies of bi-phasic catalytic reactions†
Milad
Abolhasani
a,
Nicholas C.
Bruno
b and
Klavs F.
Jensen
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 66-342, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu; Web: http://web.mit.edu/jensenlab
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA
First published on 16th April 2015
Abstract
A multi-phase flow strategy, based on oscillatory motion of a bi-phasic slug within a fluorinated ethylene propylene (FEP) tubular reactor, under inert atmosphere, is designed and developed to address mixing and mass transfer limitations associated with continuous slug flow chemistry platforms for studies of bi-phasic catalytic reactions. The technique is exemplified with C–C and C–N Pd catalyzed coupling reactions.
Over the past decade, continuous flow technologies have emerged as a powerful tool for the production of pharmaceutical targets or intermediates.1,2 However, despite the great advantages of small scale technologies compared to batch systems,3 the applicability of continuous droplet flow chemistry platforms for bi-phasic chemical reactions is limited. Slow mass transfer and mixing characteristics combined with residence time limitations, have reduced the utility of droplet-based strategies for studies of bi-phasic chemical reactions. The other challenges associated with continuous multi-phase approaches include droplet-to-droplet communication through the lubrication films surrounding the dispersed phase and solubility of the reagents or products in both phases, limiting the accuracy of the measurements. One strategy to reduce the carryover of the reagents/products between flowing droplets is adding an inert phase (e.g., inert gas, fluorinated oil) as a space between the droplets.4
The degree of mixing within the continuous and dispersed phases directly depends on the flow velocity to create the internal recirculation patterns that enhance mass transfer.5 As a result, in order to obtain similar and efficient mixing behaviors for different processing times (i.e., residence/reaction time), the length of the reactor has to be adjusted accordingly, resulting in reactor lengths exceeding 20 meters for processes with residence times longer than 20 min (with typical flow reactor inner diameter (ID) ∼1 mm and flow velocity ∼1 cm s−1).
Palladium-catalyzed C–C and C–N bond-forming reactions are two of the most widely used cross-coupling reactions in the pharmaceutical industry.6 Continuous flow technologies allow coupling reactions to be safely performed at high temperatures and/or pressures and avoid the handling of reactive or dangerous intermediates.1 While salt byproduct formation from cross-coupling reactions could potentially clog a continuous flow reactor, bi-phasic systems utilizing an aqueous base can solubilize salts, reducing the chance of clogging and allowing the reactions to proceed.7
The presence of two immiscible phases involved in the catalytic cycle necessitates a high degree of mixing and mass transfer to achieve the conversion and yield of a coupling reaction performed under high stirring rate batch conditions. Previously, Naber et al.,8 developed a continuous flow strategy for in-flow studies of bi-phasic C–N cross-coupling, using a tube reactor packed with stainless steel spheres. This approach presented an efficient strategy for studies of palladium catalyzed C–N cross-coupling, but the degree of mixing and obtained yields still depended on the volume (i.e., length) of the reactor. In addition, a solution volume of ∼1 mL was required for each experimental condition, limiting the use of this technique for high-throughput screening and optimization of bi-phasic coupling reactions. Thus, there is a need for development of a small-scale flow strategy for exploration and high-throughput screening of bi-phasic reactions, while providing similar mixing characteristics for different reactions times.
Taking advantage of the difference between the surface energies of aqueous and organic phases, we developed an oscillatory flow strategy to address the above-mentioned limitations associated with continuous flow chemistry approaches for studies of bi-phasic catalytic reactions. The new oscillatory three-phase flow reactor strategy (Fig. 1a), involves a tubular reactor (fluorinated ethylene propylene (FEP), ID 0.0625 inch), three syringe pumps, an inert gas (argon), an aqueous base, and an organic phase including the reagents, solvent and Pd precatalyst.
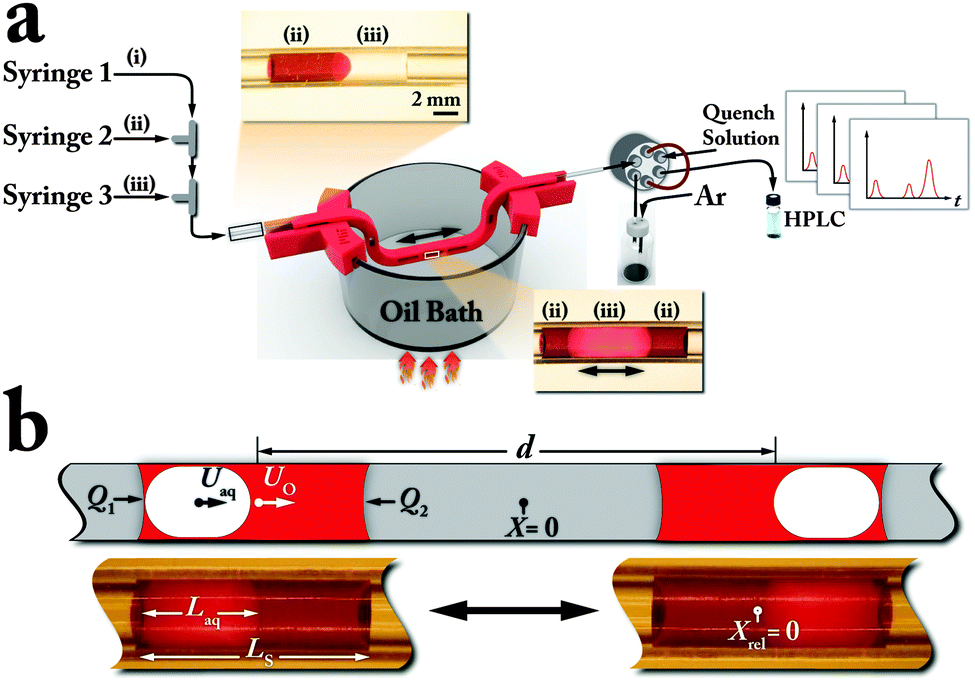 | ||
| Fig. 1 Oscillatory three-phase flow reactor. (a) Schematic of the experimental setup. Syringe 1 controls Ar, fluid (i) Syringe 2 delivers the organic phase, fluid (ii) (Sudan red dyed THF for visualization). Syringe 3 injects the aqueous base, fluid (iii) (b) Two snapshots of the engulfed aqueous droplet (clear) within the organic phase (red) at the extreme positions of the bi-phasic droplet as it oscillates. | ||
An argon (fluid (i)) filled syringe (10 psig) moves the formed droplet within the flow reactor. First, a segment of the organic phase (fluid (ii)) (5–10 μL) is formed at a T-junction, using syringe 2. The organic segment is then pushed towards the second T-junction, using syringe 1 (pressurized Ar). The aqueous base (fluid (iii)) (5–10 μL) is then injected into the organic segment, using syringe 3, to form a bi-phasic droplet. The higher surface tension of the aqueous phase compared to the organic phase of the bi-phasic droplet, in combination with the low surface energy of the fluorinated polymer tubing results in a more facile motion of the aqueous phase. Therefore, the aqueous phase of the bi-phasic droplet moves through the organic phase and leads the bi-phasic droplet (Fig. 1a top inset).
With the separation of the two phases, the available interfacial area for the catalytic reaction becomes limited to the semi-spherical interface between the organic and aqueous phases. To enhance the available interfacial area for bi-phasic catalytic reactions, it is desirable to move the aqueous phase within the organic phase of the bi-phasic droplet. Reversing the flow direction forces the aqueous phase to travel back through the organic phase (Fig. 1b and 2), and returning to the original flow makes the aqueous phase go forward again. Thus, a repeated push/pull strategy at a constant frequency with a defined flowrate, Q, and displacement volume, Vd, produces an alternating front to back motion of the aqueous droplet engulfed within the organic droplet while the bi-phasic system oscillates back and forth (Fig. 1b and 2).
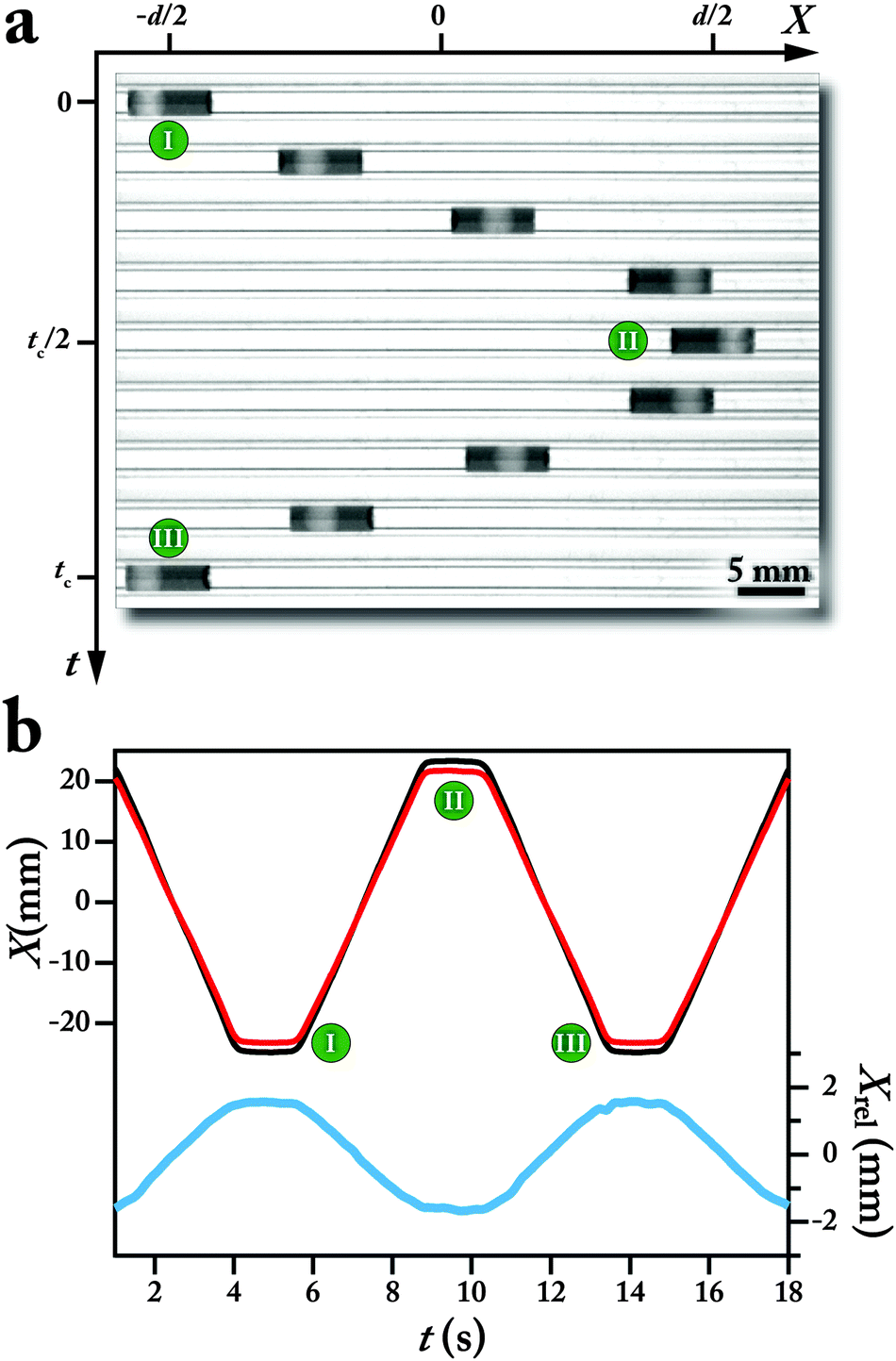 | ||
Fig. 2 (a) Bright-field micrographs of one complete oscillation cycle of a bi-phasic droplet within the tubular flow reactor (see Movie M1, ESI†). (b) Time evolution of the center point of the ( ) bi-phasic droplet and ( ) bi-phasic droplet and ( ) aqueous segment as well as the ( ) aqueous segment as well as the ( ) relative displacement of the aqueous phase with respect to the organic phase. Q = 2 mL min−1, T = 23 °C. Working fluids are argon (5 psig), dyed THF (Sudan red) and 1 M K3PO4 in DI water. The center positions of the liquid segments were obtained using 2300 images of the oscillatory motion of the bi-phasic droplet with an imaging speed of 53 frames per second. ) relative displacement of the aqueous phase with respect to the organic phase. Q = 2 mL min−1, T = 23 °C. Working fluids are argon (5 psig), dyed THF (Sudan red) and 1 M K3PO4 in DI water. The center positions of the liquid segments were obtained using 2300 images of the oscillatory motion of the bi-phasic droplet with an imaging speed of 53 frames per second. | ||
The three-phase Oscillating Flow Reactor (OFR) strategy enables the same degree of mixing for different reaction times, while removing the residence time limitation associated with continuous flow chemistry platforms. Fig. 2a shows a series snapshots of relative displacement of an aqueous droplet (1 M K3PO4 in water) within a Sudan red dyed organic phase (THF) over the course of one oscillation cycle. The absolute and relative center positions of the aqueous phase with respect to the center of the bi-phasic droplet, shown in Fig. 2b, illustrate the oscillatory motion of the aqueous droplet within the bi-phasic droplet, during one oscillation cycle.
In order to demonstrate the efficacy of the developed three-phase OFR platform for in-flow studies of palladium catalyzed C–C and C–N coupling reactions, we considered bi-phasic Suzuki–Miyaura coupling (Scheme 1a and b) and Buchwald–Hartwig amination (Scheme 1c) case studies using XPhos (3) and BrettPhos (4) precatalysts.
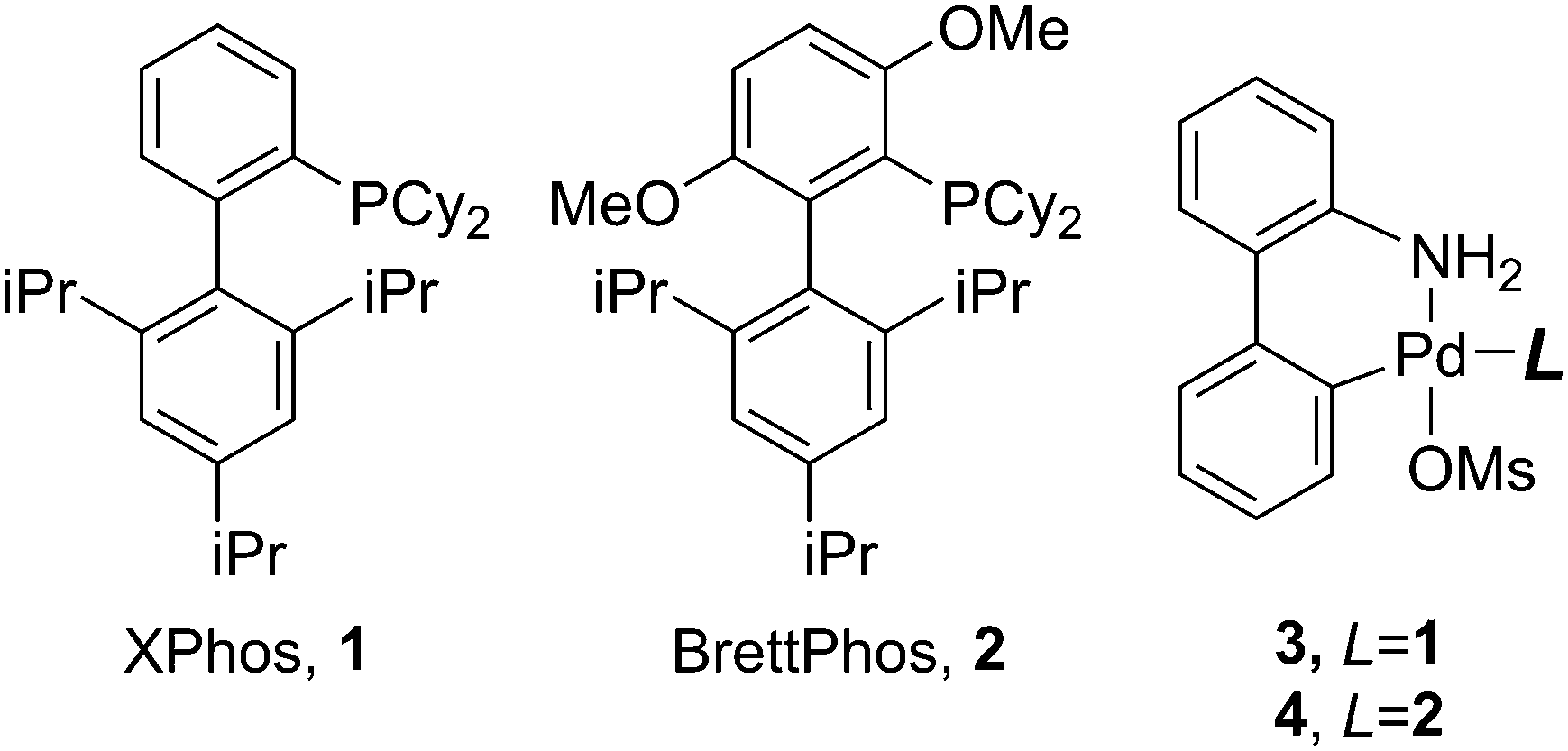
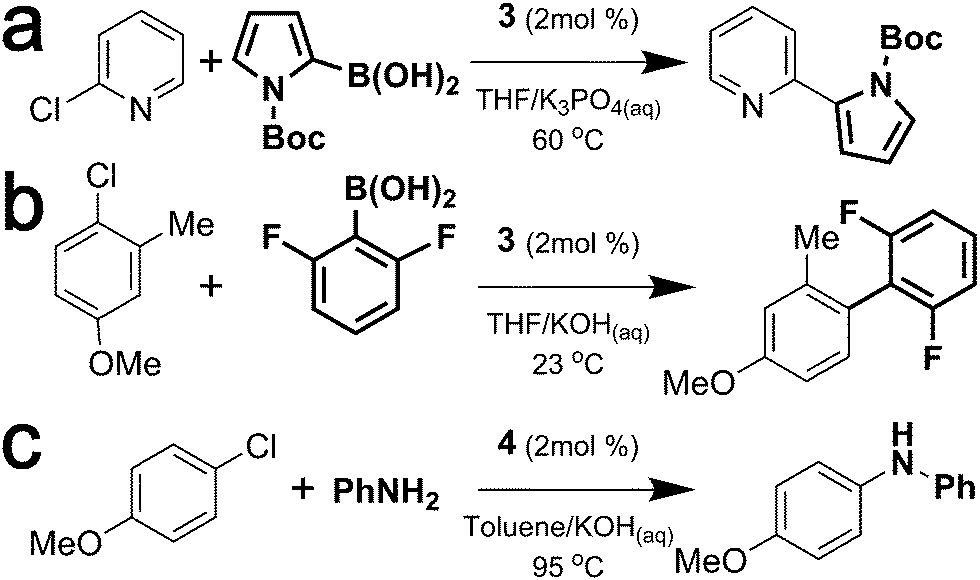 | ||
| Scheme 1 (a and b) Suzuki–Miyaura cross-couplings and (c) Buchwald–Hartwig amination used in this study. | ||
For each set of experiments, stock solutions (5 mL) for the aqueous base (stock solution A) and the organic phase (stock solution O, containing the reagents, and the Pd precatalyst dissolved in the organic solvent) were freshly prepared (see ESI†).
Batch, continuous and oscillatory flow reactions for each cross-coupling reaction (Scheme 1) were performed using the same stock solutions. Continuous flow reactor, CFR, operation of the bi-phasic Suzuki–Miyaura C–C coupling of 2-chloropyridine and N-boc-2-pyrrole boronic acid (Scheme 1a) resulted in a yield less than half of that obtained in a batch system with similar reaction conditions, whereas the OFR approach produced a similar yield to batch conditions (Fig. 3a). The lower yield obtained with CFR strategy is associated with the lower available interfacial area for the bi-phasic catalytic reaction, limiting the interactions between the aqueous base, reagents and activated Pd catalyst. In the OFR case, the moment of the aqueous droplet within the organic droplet increases the recirculation and surface area, creating conditions mirror the rapidly stirred batch reactor.
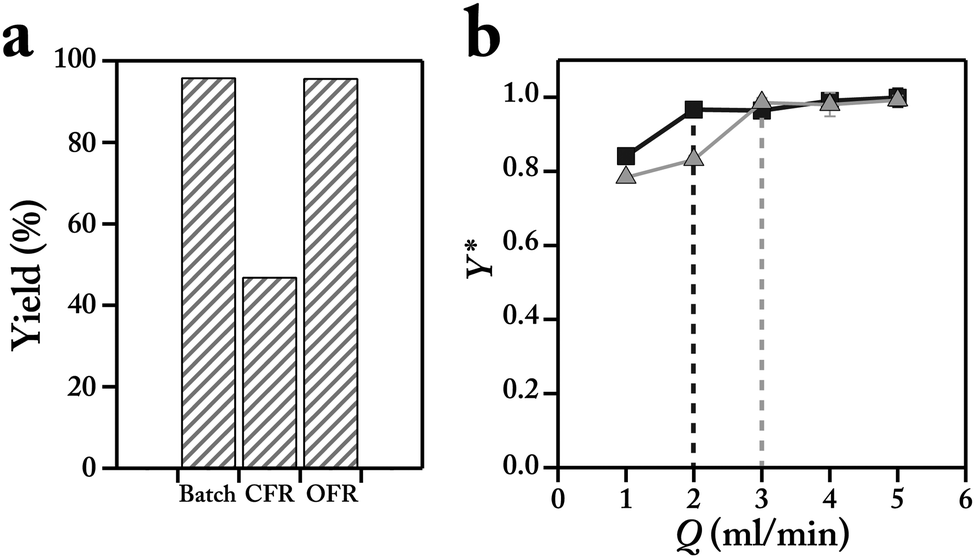 | ||
Fig. 3 (a) Comparison of the bi-phasic C–C coupling yields (Scheme 1a) between the batch, continuous and oscillatory flow reactors for the reaction time of 10 min. OFR conditions: Q = 4 mL min−1, Vd = 220 μL, and argon pressure of 7 psig. (b) Normalized OFR yields with respect to the batch yield for (■) Vd = 220 μL and ( ) Vd = 180 μL. Reaction conditions: stock solution O (5 mL), ArCl (0.25 M), boronic acid (0.375 M), internal standard, naphthalene (0.075 M) in THF. Stock solution A (5 mL), 1 M K3PO4 in water. Argon pressure for all flow experiments were set at 7 psig. Flow experiments: organic phase (10 μL), aqueous phase (5 μL). Reaction time was 10 min. Yields were obtained via HPLC and using a calibration solution of the isolated product and internal standard. The dashed lines represent the threshold flowrates for the two cases. ) Vd = 180 μL. Reaction conditions: stock solution O (5 mL), ArCl (0.25 M), boronic acid (0.375 M), internal standard, naphthalene (0.075 M) in THF. Stock solution A (5 mL), 1 M K3PO4 in water. Argon pressure for all flow experiments were set at 7 psig. Flow experiments: organic phase (10 μL), aqueous phase (5 μL). Reaction time was 10 min. Yields were obtained via HPLC and using a calibration solution of the isolated product and internal standard. The dashed lines represent the threshold flowrates for the two cases. | ||
Two parameters control the degree of mixing in the OFR strategy, (i) the total displacement volume (i.e., travel distance) of the bi-phasic droplet and (ii) the flow velocity. The latter directly correlates with the rate of recirculations inside the aqueous and organic segments. For each displacement volume, there is a threshold flowrate, Qth, where during each half cycle of the oscillatory motion, the aqueous segment completely travels from the left to the right side of the bi-phasic droplet and produces the desired interfacial contact characteristics. The threshold flow velocity is inversely correlated with the total displacement of the bi-phasic droplet. Therefore, at lower displacement volume (180 μL) the value of the threshold flowrate is larger than at the higher displacement volume (220 μL) (Fig. 3b). No significant change in the obtained coupling yields is observed for push/pull flowrates higher than 3 mL min−1, suggesting similar mixing and mass transfer characteristics for all the reactions above Qth.
The difference between the velocity of the aqueous phase (Uaq) and the overall bi-phasic droplet (Uo) is defined as the relative velocity of the aqueous phase (Urel) with respect to the organic phase. According to lubrication film theory,9,10 this relative velocity scales with the Capillary number of the aqueous phase, Caaq (Caaq = μUaq/σ, where μ is the liquid viscosity, and σ is the interfacial tension),
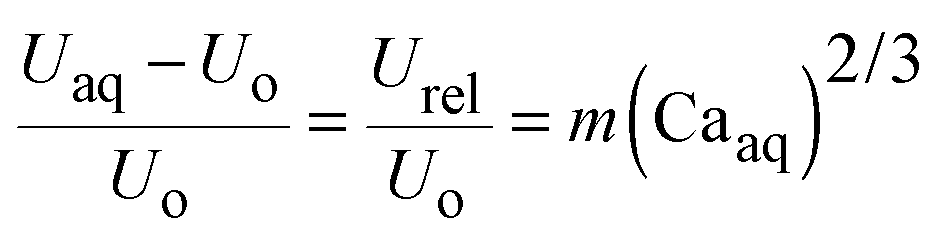 | (1) |
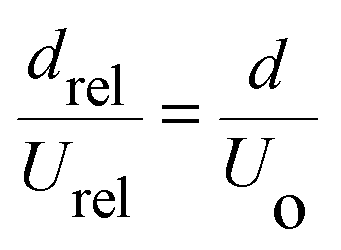 | (2) |
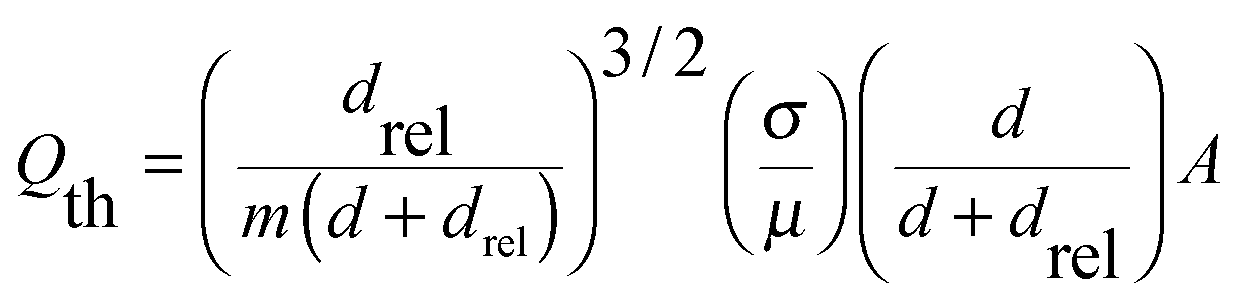 | (3) |
In order to demonstrate the versatility of the developed flow strategy, we used the developed three-phase OFR platform for in-flow Suzuki–Miyaura cross-coupling of 4-chloro-3-methylanisole and 2,6-difluorophenyl boronic acid (Scheme 1b) and Buchwald–Hartwig amination of 4-chloroanisole and aniline (Scheme 1c). As expected, the three-phase OFR platform resulted in yields similar to batch reactions for the two case studies (Fig. 4). The CFR approach always produced lower yields due to the lower available interfacial area for the bi-phasic catalytic reactions. It is noteworthy that the flowrate and the reaction time used for the Suzuki–Miyaura coupling experiment performed using the OFR approach (Fig. 4, Q = 3.5 mL min−1 and 30 min reaction time), would require a continuous reactor length of 54 m, while the OFR experiments were conducted in a 0.1 m long reactor (540 times smaller reactor volume). Furthermore, in order to increase the reactor throughput for library screening applications, multiple experiments could be conducted simultaneously in parallel tube reactors containing 1–3 bi-phasic droplets.
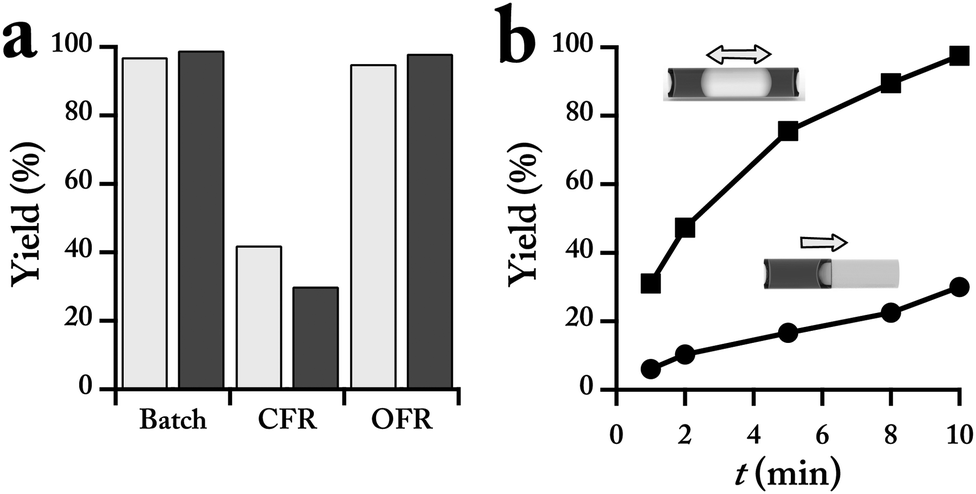 | ||
Fig. 4 Two case studies of the oscillatory flow reactor strategy: (a) ( ) Suzuki–Miyaura cross-coupling (Scheme 1b), 30 min, and (■) Buchwald–Hartwig amination (Scheme 1c), 10 min (see Movie M2, ESI†). (b) Comparison between (■) oscillatory and (●) continuous flow reactors for C–N cross-coupling reaction. Reaction conditions: (Scheme 1b) stock solution O (5 mL), ArCl (0.5 M), boronic acid (0.75 M), internal standard, naphthalene (0.1 M) in THF. Stock solution A (5 mL), 1 M KOH in water. (Scheme 1c) stock solution O (5 mL), ArCl (1 M), ArNH2 (1.2 M), internal standard, naphthalene (0.1 M) in toluene. Stock solution A (5 mL), 2 M KOH in water. Oscillatory flow conditions: Q = 3.5 mL min−1, Vd = 210 μL, organic phase (10 μL), and aqueous phase (10 μL). Argon pressure for all flow experiments were set at 10 psig. Yields were obtained via HPLC and using a calibration solution of the isolated product and internal standard. ) Suzuki–Miyaura cross-coupling (Scheme 1b), 30 min, and (■) Buchwald–Hartwig amination (Scheme 1c), 10 min (see Movie M2, ESI†). (b) Comparison between (■) oscillatory and (●) continuous flow reactors for C–N cross-coupling reaction. Reaction conditions: (Scheme 1b) stock solution O (5 mL), ArCl (0.5 M), boronic acid (0.75 M), internal standard, naphthalene (0.1 M) in THF. Stock solution A (5 mL), 1 M KOH in water. (Scheme 1c) stock solution O (5 mL), ArCl (1 M), ArNH2 (1.2 M), internal standard, naphthalene (0.1 M) in toluene. Stock solution A (5 mL), 2 M KOH in water. Oscillatory flow conditions: Q = 3.5 mL min−1, Vd = 210 μL, organic phase (10 μL), and aqueous phase (10 μL). Argon pressure for all flow experiments were set at 10 psig. Yields were obtained via HPLC and using a calibration solution of the isolated product and internal standard. | ||
In conclusion, we have developed and characterized a versatile flow chemistry platform for in-flow studies of bi-phasic catalytic reactions. In comparison with continuous flow and packed-bed approaches, the presented OFR strategy provided similar mixing and mass transfer characteristics for different reaction times without the need of adjusting the flow velocity or the length of the reactor. A total volume of 10–20 μL (200 times lower than typical batch systems) was used for each experimental condition. No “waiting time” was required for stabilization of the flow velocities or reagent concentrations. Mass transfer and residence time limitations associated with continuous flow chemistry platforms were addressed by taking advantage of the difference of surface energies between aqueous and organic solvents. Three different Pd catalyzed C–C and C–N cross-coupling reactions demonstrated the enhancement of mass transfer and mixing characteristics of the OFR approach. Yields similar to batch reactions were obtained using OFR strategy.
In addition, a guideline for selection of the appropriate flow velocity, pressure of the carrier phase and total displacement for oscillatory flow platform was provided. The developed three-phase OFR strategy could enable “small scale” flow chemistry platforms for fully automated screening, optimization, and library development of single/multi-phase chemical reactions without the need for modifying the batch chemistry, searching for a “special immiscible” carrier phase, and removing the droplet-to-droplet carryover during the course of screening experiments. The oscillatory flow approach could be utilized as a general technique in automated optimization and library developments as well as exploration of kinetics of multi-phase reactions. The resulting reaction kinetics and optimized experimental conditions could also be transferred to a scaled-up multi-phase reactor (e.g., packed-bed or batch) for continuous production of the target molecules.
We thank the Novartis Center for Continuous Manufacturing for funding this research.
Notes and references
- P. Poechlauer, J. Colberg, E. Fisher, M. Jansen, M. D. Johnson, S. G. Koenig, M. Lawler, T. Laporte, J. Manley, B. Martin and A. O'Kearney-McMullan, Org. Process Res. Dev., 2013, 17, 1472–1478 CrossRef CAS; T. Noel and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010–5029 RSC.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS PubMed; D. M. Roberge, B. Zimmermann, F. Rainone, M. Gottsponer, M. Eyholzer and N. Kockmann, Org. Process Res. Dev., 2008, 12, 905–910 CrossRef.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS PubMed; A. Gunther and K. F. Jensen, Lab Chip, 2006, 6, 1487–1503 RSC; S.-Y. Teh, R. Lin, L.-H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC; H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., 2003, 115, 792–796 CrossRef PubMed; K. S. Elvira, X. C. i Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef PubMed; X. Niu, F. Gielen, J. B. Edel and A. J. deMello, Nat. Chem., 2011, 3, 437–442 CrossRef PubMed; R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef PubMed; A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef PubMed.
- J. E. Kreutz, A. Shukhaev, W. Du, S. Druskin, O. Daugulis and R. F. Ismagilov, J. Am. Chem. Soc., 2010, 132, 3128–3132 CrossRef CAS PubMed; B. Zheng and R. F. Ismagilov, Angew. Chem., Int. Ed., 2005, 44, 2520–2523 CrossRef PubMed; S. Duraiswamy and S. A. Khan, Nano Lett., 2010, 10, 3757–3763 CrossRef PubMed; A. B. Theberge, G. Whyte, M. Frenzel, L. M. Fidalgo, R. C. R. Wootton and W. T. S. Huck, Chem. Commun., 2009, 6225–6227 RSC.
- V. Sebastian Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef CAS PubMed; B. K. H. Yen, A. Günther, M. A. Schmidt, K. F. Jensen and M. G. Bawendi, Angew. Chem., 2005, 117, 5583–5587 CrossRef PubMed; J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef.
- F. W. Goldberg, R. A. Ward, S. J. Powell, J. É. Debreczeni, R. A. Norman, N. J. Roberts, A. P. Dishington, H. J. Gingell, K. F. Wickson and A. L. Roberts, J. Med. Chem., 2009, 52, 7901–7905 CrossRef CAS PubMed; J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef PubMed; C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef PubMed; K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef PubMed; C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef PubMed.
- M. Carril, R. SanMartin and E. Dominguez, Chem. Soc. Rev., 2008, 37, 639–647 RSC; B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS PubMed; K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef PubMed.
- J. R. Naber and S. L. Buchwald, Angew. Chem., 2010, 122, 9659–9664 CrossRef PubMed.
- L. W. Schwartz, H. M. Princen and A. D. Kiss, J. Fluid Mech., 1986, 172, 259–275 CrossRef CAS; F. P. Bretherton, J. Fluid Mech., 1961, 10, 166–188 CrossRef.
- T. C. Thulasidas, M. A. Abraham and R. L. Cerro, Chem. Eng. Sci., 1995, 50, 183–199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental setup, image processing and product characterization methods. See DOI: 10.1039/c5cc02051d |
| This journal is © The Royal Society of Chemistry 2015 |