
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically and electrochemically induced expansion and contraction of a ferrocene rotor†
Synøve Ø.
Scottwell
a,
Anastasia B. S.
Elliott
ab,
Karl J.
Shaffer
a,
Ayman
Nafady
c,
C. John
McAdam
a,
Keith C.
Gordon
ab and
James D.
Crowley
*a
aDepartment of Chemistry, University of Otago, P.O. Box 56, Dunedin, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz; Fax: +64 3 479 7906; Tel: +64 3 479 7731
bMacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
cDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 1st April 2015
Abstract
A 2,2′-bipyridine-appended ferrocene rotor, 1,1′-di(5-yl-ethynyl-2,2′-bipyridine)ferrocene, can be switched from a folded/stacked (syn) conformation to an extended/unstacked (anti) conformation by the addition of [Cu(CH3CN)4](PF6) and 6,6′-dimesityl-2,2′-bipyridine. This extension and contraction process was completely reversible and could be triggered either chemically or electrochemically.
All the cellular processes of life are carried out by biological (nano)molecular machines.1 Inspired by these systems chemists have begun to develop switchable synthetic molecular machines.2 While there have been some spectacular successes, including synthetic rotary motors,3 molecular muscles,4 and sequence-specific peptide synthesis machines5 the vast majority of synthetic systems developed are based on complex and difficult to synthesise mechanically interlocked architectures (MIAs).6 The challenging and laborious synthesis of these MIAs has led to increasing efforts to develop potentially synthetically more accessible non-interlocked molecular machines.7 In particular systems incorporating metal ions are becoming more and more common due to the favourable kinetic and thermodynamic properties of the coordination bonds.8
The reversible electrochemistry9 and molecular ball-bearing10 properties of ferrocene (Fc) derivatives11 have made them useful building blocks for the development of non-interlocked synthetic molecular machines.12 We and others have exploited ferrocene to develop electrostatically driven two-state switches (Fig. 1).13 We have shown that pyridyl and 2,2′-bipyridyl (bpy) 1,1′-disubstituted ferrocenes13c initially adopt the stacked syn-conformation due to the electronic preference of the ferrocene and favourable π–π interactions. Introduction of charge to the ferrocene “arms”, either by protonation or metallation with Pd2+ ions, induced rotation to the anti-conformation to minimise the electrostatic repulsion between the cationic substituents of the ferrocene rotors.13c Deprotonation or addition of chloride ions removed the electrostatic repulsion restoring the initial stacked syn conformation.13c Other workers13a,b have generated electrochemical switchable systems by incorporating redox-active organic arms into the ferrocene rotors.
 | ||
| Fig. 1 Generic stimuli responsive switching with ferrocene (Fc) rotor molecules and the compounds 1a–b and 2a–b studied herein. | ||
Herein we build on our previous work13c and develop ferrocene rotor systems that can be switched from the syn to anti conformation by the complexation/decomplexation of a copper 6,6′-dimesityl-2,2′-bipyridine fragment. A combination of electrostatic and steric repulsion destabilises the syn conformation upon complexation generating the anti rotamer. This extension and contraction process was completely reversible and could be triggered either chemically or electrochemically.
The ethynylferrocene rotors (1a–b) and model (2a–b) systems were readily synthesised using modified literature procedures (ESI†).141H NMR spectroscopy, density functional theory (DFT) calculations and X-ray crystallography were used to determine the preferred conformation of the ferrocene rotors (1a–b). The proton signals associated with the bipyridyl and pyridyl rings (Hd–j) of 1a–b were shifted upfield relative to those of the singly armed model compounds 2a–b, indicating that the di-armed ferrocene rotors adopt a stacked (syn) conformation in solution (Fig. 2a and b and ESI,† Fig. S38). This was further supported by computational modelling of the systems. DFT calculations (CAM-B3LYP, DMF solvent field)13b,15 were used to determine the relative energies of a series of different rotation conformers of 1a–b (the angles between the ferrocene “arms” of 1a–b was varied from −180° to 180° and the compound energy minimised). These calculations showed, as expected, that the stacked, fully eclipsed (syn) rotamers (angle = 0°) were the lowest energy conformations for both 1a and 1b by 3 and 6 kJ mol−1, respectively (ESI,† Fig. S82 and S86). Additional evidence for the stacked (syn) conformation was obtained from X-ray crystallography (Fig. 3a and ESI†). The molecular structure of 1a clearly showed that the bipyridine substituents are stacked (centroid–centroid distances 3.636 and 3.716 Å) and the ferrocene cyclopentadiene (Cp) rings are eclipsed, with a dihedral angle of 1.58° (Fig. 3a) between the substituents consistent with the 1H NMR spectral and computational data.
 | ||
| Fig. 2 1H NMR spectra (400 MHz, CDCl3, 298 K) of (a) 2a (b) 1a (c) the copper(I) complex [Cu2(1a)(3)2](PF6)2 (d) cyclam induced de-metallation/switching (e) addition of 2 eq. of [Cu(CH3CN)4](PF6) (f) cyclam induced de-metallation/switching. | ||
 | ||
| Fig. 3 ORTEP18 diagrams showing the solid state structures of the rotor 1a (a), and the Cu(I) complex [Cu2(1a)(3)2](PF6)2 (b). The ellipsoids are shown at the 50% probability level. Counterions and hydrogen atoms of the complex are omitted for clarity. Additional crystallographic details can be found in the ESI.† | ||
Reaction of either 1a, 1b or 2a in acetonitrile (CH3CN) solution with [Cu(CH3CN)4](PF6) and the sterically bulky bipyridine ligand 6,6′-dimesityl-2,2′-bipyridine16 (3) was accompanied by a colour change from yellow/orange to red/brown indicative of complexation (Scheme 1(i) and ESI†).171H NMR spectroscopy (Fig. 2c and ESI,† Fig. S41 and S42) and ESMS (ESI† Fig. S64–S67) confirmed the formation of the expected copper(I) complexes. Due to the combination of electrostatic and steric repulsion generated upon complex formation it was expected that the ferrocene rotor units (1a and 1b) would switch conformation from the syn to the anti rotamer. An examination of the 1H NMR spectra of the complex [Cu(1b)(3)](PF6)2, the rotor 1b and the corresponding model compound 2b provided experimental support for this syn to anti conformation switch on complexation. As discussed above, the proton resonances of the methylpyridinum unit of 1b are shifted upfield relative to those of the model compound 2b, indicating that the di-armed ferrocene rotor adopts a stacked (syn) conformation in solution (ESI,† Fig. S38). Upon complexation to the copper(I) 3 complex the proton signals of the methylpyridinum unit shift downfield to values that are almost identical to those observed from the model compound 2b indicating that the pyridinum arm of the copper(I) complex is no longer π-stacked (ESI,† Fig. S41). DFT calculations (CAM-B3LYP, DMF solvent field)13b,15 were used to examine the relative energies of different rotational conformers of the copper(I) complexes [Cu2(1a)(3)2](PF6)2, and [Cu(1b)(3)](PF6)2 (ESI,† Fig. S84 and S88). The resulting plot revealed that the minimum angle between the two arms could not be less than 60°, with the energy difference between higher angle conformations being negligible. Thus it can be surmised that the arms of the rotor must have extended to at least 60° in solution. Consistently, the molecular structure of [Cu2(1a)(3)2]2+ as the perchlorate (ClO4−) salt (Fig. 3b and ESI†) showed that the [Cu(3)]+ units had coordinated to both bipyridine arms of 1a in the expected “pacman-like” arrangement. More importantly it confirmed that the rotor molecule was almost completely extended, with the angle between the two arms ranging from 142–145° (Fig. 3b and ESI†).
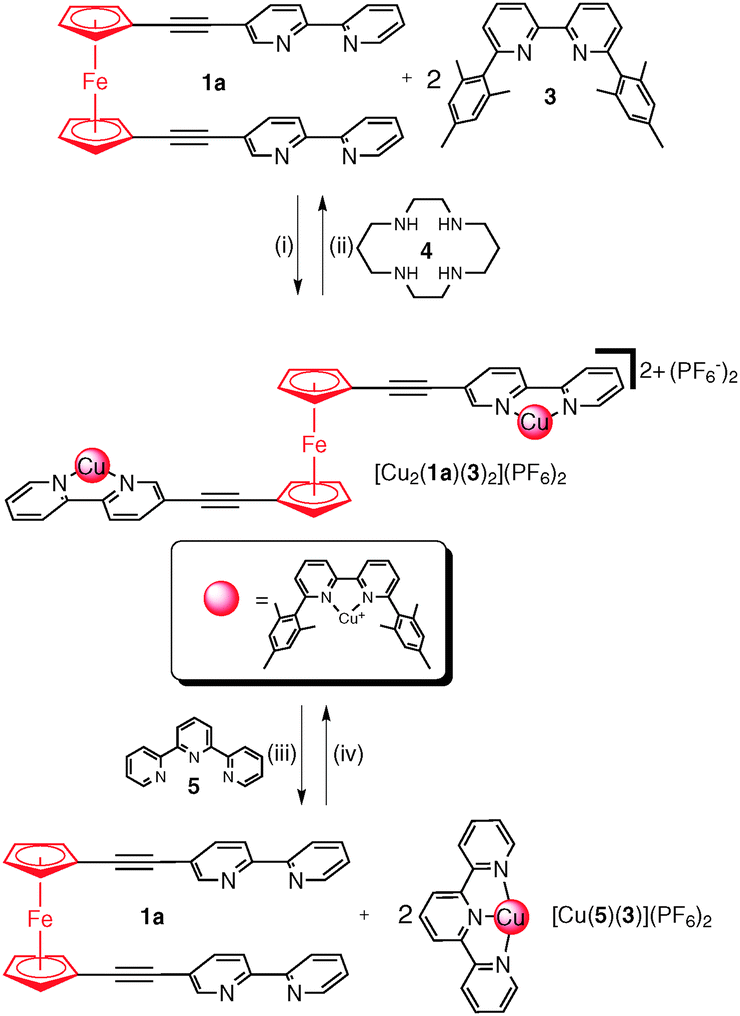 | ||
| Scheme 1 Synthesis of the copper(I) complex of 1a and the chemical and electrochemical switching. (i) [Cu(CH3CN)4](PF6) (2 eq.), CDCl3, 298 K; (ii) cyclam 4 (2 eq.), CDCl3, 298 K; (iii) 2,2′:6′,2′′-terpyridine (5) (2 eq.), −2e−, acetone, 0.1 M NBu4PF6; and (iv) +2e−, acetone, 0.1 M NBu4PF6. | ||
Having confirmed that the rotors 1a–b (syn) and the corresponding copper(I) complexes (anti) adopt two different conformations we next examined if we could cleanly and reversibly switch between the rotor (1a) and rotor complex [Cu2(1a)(3)](PF6)2. Addition of two equivalents of 1,4,8,11-tetraazacyclotetradecane (cyclam, 4) to an acetone solution of [Cu2(1a)(3)2](PF6)2 caused an immediate colour change from red to orange (Scheme 1(ii), Fig. 2c and d and ESI†), indicating that the Cu(I) ions had been removed from the ferrocene complex.191H NMR and UV-vis spectroscopy and ESMS (Fig. 2c and d and ESI†) confirmed that only the free rotor 1a and free 6,6′-dimesityl-2,2′-bipyridine ligand 3 were present in solution. Addition of two equivalents of [Cu(CH3CN)4](PF6) to the solution regenerated the characteristic red colour of the complex [Cu2(1a)(3)2](PF6)2, and the 1H NMR spectrum (Fig. 2e) indicated that the complex had quantitatively re-formed. This chemically driven syn-to-anti switching process could be repeated a number of times (>5) indicating that switching was completely reversible (ESI,† Fig. S47). However, the method led to the build-up of waste by-products (Cu(II)-cyclam) which is uneconomical and could potentially lead to the loss of function.20 Therefore we explored alternative switching methodologies. The electrochemical Cu(I)/Cu(II) switching process developed by Sauvage et al.21 for the generation of a range of MIAs molecular switches and machines seemed ideal.22
The CV (100 mV s−1, acetone, 0.1 M NBu4PF6) of the [Cu2(1a)(3)2](PF6)2 rotor complex displays two chemically reversible processes at 0.64 and 0.85 V respectively (Fig. 4 red/brown trace). By comparison with the previously prepared [(3-bpyFc)Cu(3)](PF6)17 and [Cu(3)(bpy)](BF4)23 model systems the first oxidation process was assigned to the CuII/I couple while the second oxidation process is ferrocenyl based.24 These assignments were further supported by two-step bulk electrolysis experiments. A stepwise oxidation was performed on the [Cu2(1a)(3)2](PF6)2 complex, oxidation at the potential of the first wave (Eappl = 0.72 V) passed 2.0 F mol−1 with the oxidized species [Cu2(1a)(3)2]4+ proving stable at ambient temperature. The second step at Eappl = 1.1 V passed only 1.0 F mol−1, thereby confirming the transfer of two and one electrons, respectively. These results confirmed that the first process is the Cu(I) to Cu(II) oxidation while the second is the oxidation of the ferrocene to ferrocenium.
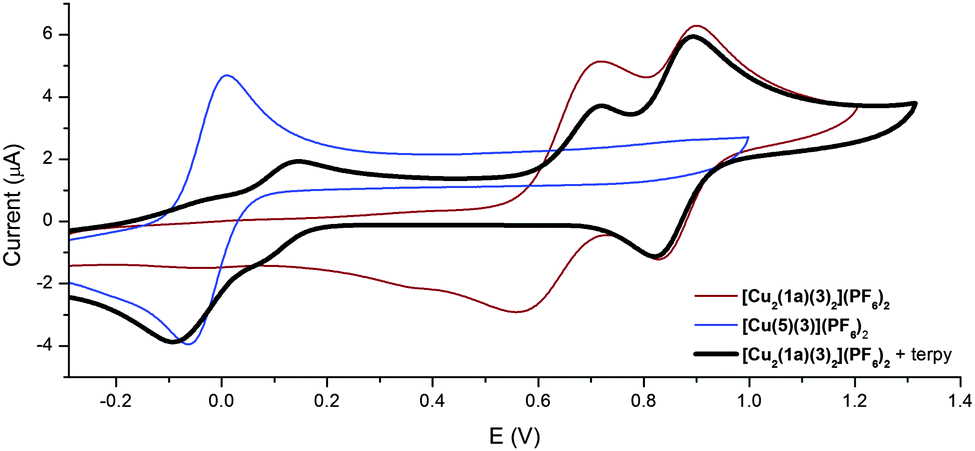 | ||
| Fig. 4 CVs (100 mV s−1) of the Cu(I) complex [Cu2(1a)(3)2](PF6)2, the Cu(II) terpy complex [Cu(5)(3)](PF6)2, and [Cu2(1a)(3)2](PF6)2 in the presence of terpy in acetone. | ||
The CV experiment was then carried out in the presence of two equivalents of 2,2′:6′,2′′-terpyridine (5). The anodic potential sweep showed the two peaks corresponding to the CuII/I and ferrocenyl oxidation processes. On the reverse scan, only the reduction of the ferrocenium back to ferrocene is observed. No peak corresponding to the reduction of the [Cu2(1a)(3)2]4+ complex was observed. Instead proceeding in the cathodic direction, a peak corresponding to the reduction of the pentacoordinated [Cu(5)(3)](PF6)2 complex was observed at −0.10 V (Fig. 4 black trace). This electrochemical behaviour is very similar to that observed by Sauvage et al.21 in their MIAs systems.
Bulk electrolysis in the presence of two equivalents of 2,2′:6′,2′′-terpyridine (5) was accompanied by a colour change from deep red/brown to yellow, the same as was observed for the chemical switching process with cyclam (ESI,† Fig. S81). Spectroelectrochemistry (acetone, 0.1 M Bu4NPF6, OTTLE cell, ESI,† Fig. S80) of the [Cu2(1a)(3)2](PF6)2 complex confirmed that during the bulk oxidation the MLCT band (λmax = 476 nm) decreases until a spectrum consistent with “free” 1a is generated. ESI-MS analysis of the 2,2′:6′,2′′-terpyridine (5) and [Cu2(1a)(3)2](PF6)2 mixture before bulk electrolysis displayed peaks due to the [Cu2(1a)(3)2(PF6)]+, [Cu2(1a)(3)2]2+ and [5 + H]+ ions. After oxidation the mass spectrum only contain peaks consistent with [Cu(5)(3)]2+, [1a + H]+ and [1a + Na]+ ions.
Combined these results suggest that, upon oxidation or reduction of the copper ions, the [Cu(3)]n+ fragments are set in motion. Upon oxidation of [Cu2(1a)(3)2]2+ to [Cu2(1a)(3)2]4+, the resulting tetrahedrally coordinated Cu(II) ions are unstable and this leads to the migration of the [Cu(3)]n+ fragment to the 2,2′:6′,2′′-terpyridine (5) ligands generating the more stable five-coordinate [Cu(5)(3)]2+ complex (Scheme 1(iii)). Similarly, upon reduction of the Cu(II) back to Cu(I) the [Cu(5)(3)]+ complex is unstable as the copper(I) ions are in the pentacoordinate trigonal bipyramidal environment. As such the [Cu(3)]n+ fragments migrate back to the ferrocene rotor 1a regenerating the complex [Cu2(1a)(3)2]2+ and returning the copper(I) ions to the preferred tetrahedral coordination environment.
Herein we have shown that readily synthesised 2,2′-bipyridine-appended ferrocene rotor molecules can be switched from a folded/stacked (syn) to an extended/unstacked (anti) conformation upon the addition of [Cu(CH3CN)4](PF6) and 6,6′-dimesityl-2,2′-bipyridine to the rotor units. This switching is driven by a combination of electrostatic and steric repulsion which destabilizes the syn conformation upon copper(I) complexation and generates the anti rotamer. This extension and contraction process was completely reversible and could be triggered either chemically or electrochemically. The chemically driven process can be repeated multiple times but a large amount of Cu(II)-cyclam waste slowly builds up in solution. The switching of the rotor can also be triggered electrochemically. Oxidative bulk electrolysis of the [Cu2(1a)(3)2](PF6)2 rotor complex in the presence of two equivalents of 2,2′:6′,2′′-terpyridine (5) was accompanied by a colour change from deep red/brown to yellow indicative of the translocation of the [Cu(3)]2+ fragment to the 2,2′:6′,2′′-terpyridine (5) ligands and the formation of syn-1a. Reduction of the mixture restored the original red/brown colour suggesting the [Cu(3)]+ fragment has moved from the 2,2′:6′,2′′-terpyridine (5) back to the rotor 1a regenerating the extended anti-[Cu2(1a)(3)2](PF6)2 rotor complex. This electrochemical process provides a clean reversible method for the extension and contraction of the ferrocene rotors.
The findings presented here could enable the generation of new, readily accessible, non-interlocked, electrochemically switchable bistable molecular machines or catalysts.25 Inspired by the molecular “folding ruler” of Takeuchi and co-workers26 we are currently attempting to generate longer ferrocene oligomers that will undergo a larger molecular extension and contraction and exploit these molecules as (nano)molecular actuators.
SØS and ABSE thank the University of Otago for a PhD scholarship. CJM thanks the NZ Ministry of Business, Innovation and Employment Science Investment Fund (Grant no. UOOX1206) for financial support. AN thanks Scientific Research at King Saud University for funding (RGP-VPP-236). JDC thanks the Marsden Fund (Grant no. UOO1124) and the University of Otago (Division of Sciences Research Grant) for supporting this work.
Notes and references
- M. Schliwa, Molecular Motors, Wiley-VCH Verlag GmbH & Co. KGaA, 2003 Search PubMed.
- (a) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed; (b) S. Silvi, M. Venturi and A. Credi, Chem. Commun., 2011, 47, 2483–2489 RSC; (c) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; (d) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- (a) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed; (b) J. V. Hernandez, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef CAS PubMed.
- (a) G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed; (b) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- (a) G. Barin, R. S. Forgan and J. F. Stoddart, Proc. R. Soc. A, 2012, 468, 2849–2880 CrossRef CAS PubMed; (b) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- (a) For some reviews see; (b) P. C. Knipe, S. Thompson and A. D. Hamilton, Chem. Sci., 2015, 6, 1630–1639 RSC; (c) M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656–3676 RSC; (d) G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473–487 CrossRef CAS PubMed.
- (a) For some selected recent examples see; (b) A. Noor, D. L. Maloney, J. E. M. Lewis, W. K. C. Lo and J. D. Crowley, Asian J. Org. Chem., 2015, 4, 208–211 CrossRef CAS PubMed; (c) J. E. Beves, V. Blanco, B. A. Blight, R. Carrillo, D. M. D'Souza, D. Howgego, D. A. Leigh, A. M. Z. Slawin and M. D. Symes, J. Am. Chem. Soc., 2014, 136, 2094–2100 CrossRef CAS PubMed; (d) N. Zigon, P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2014, 50, 5040–5042 RSC; (e) D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757–762 CrossRef CAS PubMed; (f) D. Sooksawat, S. J. Pike, A. M. Z. Slawin and P. J. Lusby, Chem. Commun., 2013, 49, 11077–11079 RSC; (g) S. J. Pike and P. J. Lusby, Chem. Commun., 2010, 46, 8338–8340 RSC.
- U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S. W. Hla, Nat. Nanotechnol., 2013, 8, 46–51 CrossRef CAS PubMed.
- C. Li, J. C. Medina, G. E. M. Maguire, E. Abel, J. L. Atwood and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 1609–1618 CrossRef CAS.
- (a) K. Nikitin, H. Muller-Bunz, Y. Ortin, J. Muldoon and M. J. McGlinchey, J. Am. Chem. Soc., 2010, 132, 17617–17622 CrossRef CAS PubMed; (b) T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS PubMed; (c) K. Heinze and M. Schlenker, Eur. J. Inorg. Chem., 2005, 66–71 CrossRef CAS PubMed; (d) X.-B. Wang, B. Dai, H.-K. Woo and L.-S. Wang, Angew. Chem., Int. Ed., 2005, 44, 6022–6024 CrossRef CAS PubMed; (e) T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612–5613 CrossRef CAS PubMed.
- (a) Metallacarboranes have also been used to develop similar molecular rotors, see; (b) M. F. Hawthorne, J. I. Zink, J. M. Skelton, M. J. Bayer, C. Liu, E. Livshits, R. Baer and D. Neuhauser, Science, 2004, 303, 1849–1851 CrossRef CAS PubMed; (c) A. V. Safronov, N. I. Shlyakhtina, T. A. Everett, M. R. VanGordon, Y. V. Sevryugina, S. S. Jalisatgi and M. F. Hawthorne, Inorg. Chem., 2014, 53, 10045–10053 CrossRef CAS PubMed.
- (a) A. Takai, T. Yasuda, T. Ishizuka, T. Kojima and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 9167–9171 CrossRef CAS PubMed; (b) A. Iordache, M. Oltean, A. Milet, F. Thomas, B. Baptiste, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2012, 134, 2653–2671 CrossRef CAS PubMed; (c) J. D. Crowley, I. M. Steele and B. Bosnich, Chem. – Eur. J., 2006, 12, 8935–8951 CrossRef CAS PubMed.
- (a) M. S. Inkpen, S. Du, M. Driver, T. Albrecht and N. J. Long, Dalton Trans., 2013, 42, 2813–2816 RSC; (b) M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Chem. Commun., 2013, 49, 5663–5665 RSC.
- W.-Y. Wang, N.-N. Ma, S.-L. Sun and Y.-Q. Qiu, Phys. Chem. Chem. Phys., 2014, 16, 4900–4910 RSC.
- M. Schmittel, A. Ganz, W. A. Schenk and M. Hagel, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 559–564 CAS.
- S. Ø. Scottwell, K. J. Shaffer, C. J. McAdam and J. D. Crowley, RSC Adv., 2014, 4, 35726–35734 RSC.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- (a) M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832–3836 CrossRef CAS PubMed; (b) G. Haberhauer, Angew. Chem., Int. Ed., 2008, 47, 3635–3638 CrossRef CAS PubMed.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef CAS PubMed.
- (a) N. Armaroli, V. Balzani, J.-P. Collin, P. Gavina, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397–4408 CrossRef CAS; (b) A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS.
- (a) During the course of our work some other non-interlocked systems that exploit the Cu(I)–Cu(II) redox switching method of Sauvage and co-workers have been developed, see; (b) F. Niess, V. Duplan and J.-P. Sauvage, J. Am. Chem. Soc., 2014, 136, 5876–5879 CrossRef CAS PubMed; (c) S. K. Samanta, A. Rana and M. Schmittel, Dalton Trans., 2014, 43, 9438–9447 RSC; (d) S. Pramanik, S. De and M. Schmittel, Chem. Commun., 2014, 50, 13254–13257 RSC.
- M. G. Fraser, H. van der Salm, S. A. Cameron, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2013, 52, 2980–2992 CrossRef CAS PubMed.
- The electrochemical properties of the ligands and complexes were determined using a combination of cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments (ESI†).
- (a) S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed; (b) M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730–11732 RSC; (c) V. Blanco, A. Carlone, K. D. Haenni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- M. Shibata, S. Tanaka, T. Ikeda, S. Shinkai, K. Kaneko, S. Ogi and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 397–400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR, HR-ESI-MS, UV-vis, electrochemical and crystallographic data. CCDC 1046336, 1046337 and 1046339–1046342. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01973g |
| This journal is © The Royal Society of Chemistry 2015 |