
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed tandem phosphination–decarboxylation–oxidation of alkynyl acids with H-phosphine oxides: a facile synthesis of β-ketophosphine oxides†
Pengbo
Zhang
a,
Liangliang
Zhang
a,
Yuzhen
Gao
a,
Jian
Xu
a,
Hua
Fang
b,
Guo
Tang
*a and
Yufen
Zhao
a
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, and the Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen, Fujian 361005, China. E-mail: t12g21@xmu.edu.cn; Fax: +86-592-2185780
bThird Institute of Oceanography, State Oceanic Administration, Xiamen, Fujian 361005, China
First published on 31st March 2015
Abstract
The general method for the tandem phosphination–decarboxylation–oxidation of alkynyl acids under aerobic conditions has been developed. In the presence of CuSO4·5H2O and TBHP, the reactions provide a novel access to β-ketophosphine oxides in good to excellent yields. This transformation allows the direct formation of a P–C bond and the construction of a keto group in one reaction.
Organophosphorus compounds have broad applications in the fields of organic synthesis,1 materials science,2 medicinal chemistry,3 and ligand chemistry.4 Thus, development of a new efficient method for C–P bond construction has attracted increasing attention. β-Ketophosphine oxides can facilitate carbon–carbon bond formation and then the diphenylphosphinyl group can be easily removed to give olefins,5 cyclopropanes,6 and branched ketones.7 β-Ketophosphine oxides can also be used for liquid–liquid extraction of metal ions because of their prominent metal-complexing abilities.8 The traditional methods to prepare β-ketophosphine oxides are based on the acylation of alkyl phosphine oxides with carboxylic acid derivatives which employ stoichiometric amounts of the hazardous organometallic reagents.9 In recent years, our group10 and other researchers11 reported many practical approaches to β-ketophosphonates, but these methods are not ideal choices for the synthesis of β-ketophosphine oxides.
In 1966, Nilsson reported the pioneering work of decarboxylative coupling.12 Since 2002, a series of transition-metal-catalyzed decarboxylative C–C and C–heteroatom bond formation reactions have been extensively developed.13 Compared with the traditional cross-coupling reactions and C–H activation, decarboxylative coupling reactions using carboxylic acid derivatives have several advantages. Instead of metal waste from organometallic coupling reagents, less toxic carbon dioxide is released as a byproduct after the complete conversion, which reduces the cost of the process for the treatment of waste. It is noteworthy that as a practical alternative, the use of arylpropiolic acids as terminal arylacetylene surrogates is safer and more attractive because arylpropiolic acids are usually solids without an unpleasant smell and are convenient to synthesize, store, and transport. On the basis of this viewpoint, Wu's group fulfilled the decarboxylative coupling of arylpropiolic acids with P(O)H to construct a Csp–P bond with the assistance of a copper catalyst system.14 Recently, our group developed an efficient synthesis of E-alkenylphosphine oxides via copper-catalyzed decarboxylative cross-coupling of alkynyl acids with H-phosphine oxides.15 To the best of our knowledge, the example of β-ketophosphine oxide formation via decarboxylative coupling of alkynyl acids is yet to be reported (Scheme 1).
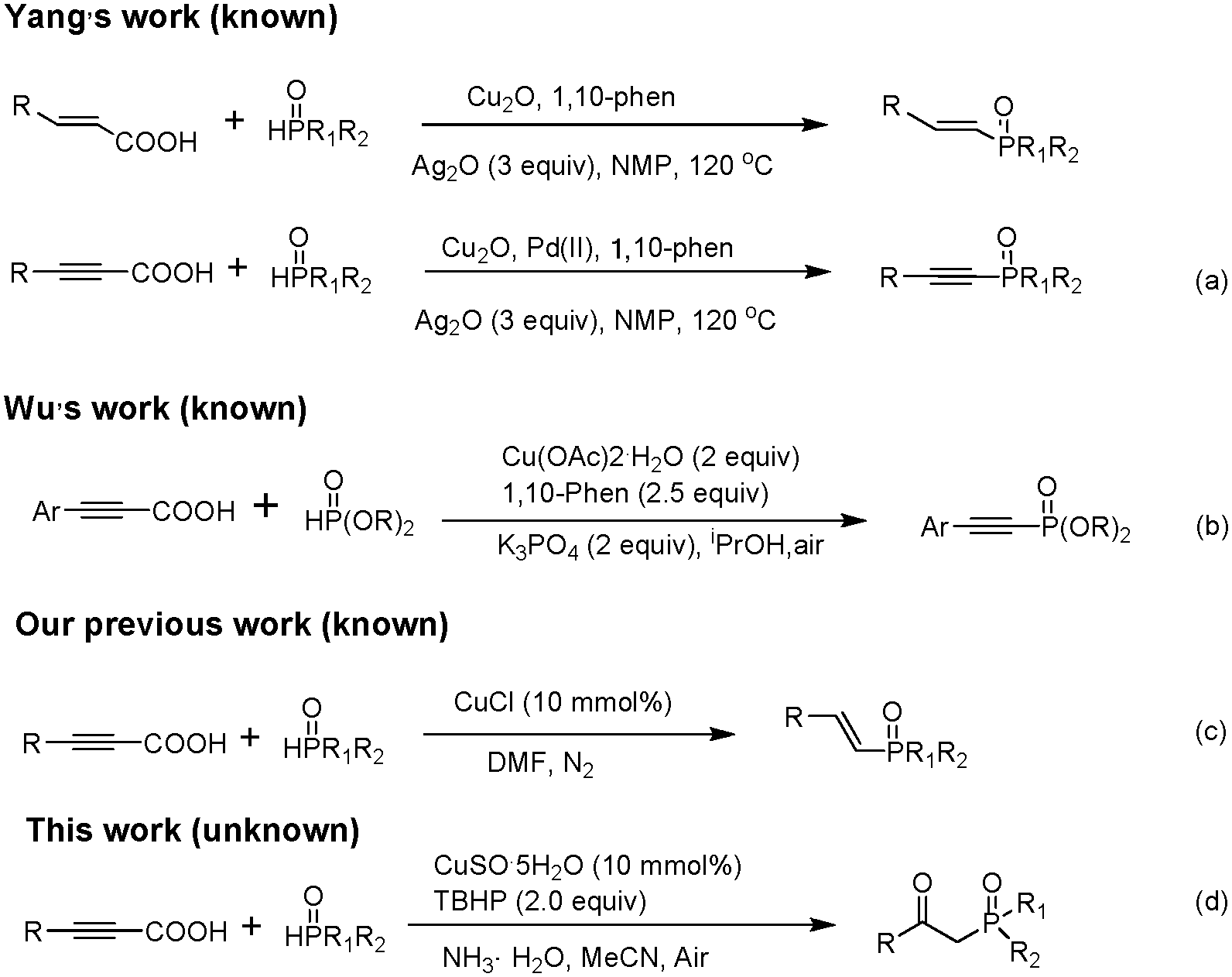 | ||
| Scheme 1 C–P bond formation via decarboxylative coupling. | ||
Reactions involving organophosphorus radicals have a long history, and are useful reactive species in organic synthetic chemistry.16 Owing to our continuous interest in the P–C bond formation17 and the reaction of organophosphorus radicals,18 we present herein our recent progress in constructing valuable β-ketophosphine oxides via tandem phosphination–decarboxylation–oxidation of alkynyl acids. This transformation allows the direct formation of a P–C bond and the construction of a keto group in one reaction via a radical process.
At the outset of our investigation, phenylpropiolic acid (1a) and H(O)PPh2 (2a) were chosen as the model substrates to survey the reaction conditions. Gratifyingly, when a mixture of 1a (0.2 mmol), 2a (0.8 mmol), CuSO4·5H2O (0.02 mmol) and NH3·H2O (25%, 0.25 mL) in MeCN was heated to 60 °C in air for 2 h, the desired product 3a was obtained in 43% yield (Table 1, entry 1). Subsequently, various Cu(I) and Cu(II) salts were further checked and the results showed that Cu(II) salts were more effective to give the desired product (entries 1–9). A brief survey of bases such as Cs2CO3, K2CO3, NaOAc, (iPr)2NEt, NEt3, pyridine, and NH3·H2O (25%) led to the observation that NH3·H2O (25%) gave 3a in the highest yield (entries 9–16). In our previous synthesis of α-hydroxy phosphonates from H-phosphonates and alcohols, we found that the combined use of Cu(II) and TBHP (tert-butylhydroperoxide) could promote the reaction efficiently.16h Gratifyingly, the yield increased tremendously when TBHP was employed as an oxidant. However, the other oxidants like K2S2O8, BQ (p-benzoquinone), DTBP (di-tert-butyl peroxide), and H2O2 did not improve the yield (entries 17–22). The solvent systems employed also notably affected the related reaction efficiencies. Conducting the reaction in EtOH, DMF, DMSO and 1,4-dioxane gave the product 3a in very low yield (entries 23–26), while the reaction conducted in MeCN gave a high yield (entry 18). Moreover, the yield was reduced to 30% when using O2 instead of TBHP (entry 22). No desired product was afforded without copper salts (entry 27). The yield of product 3a decreased when the temperature was raised to 70 °C or decreased to room temperature (entry 28). However, the attempt to decrease the amount of 2a failed (entry 29). After optimization of the reaction conditions, we established a highly efficient route to the tandem decarboxylation–phosphination–oxidation of alkynyl acids (entry 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Baseb | Solvent | Oxidant | Yieldc (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.8 mmol), catalyst (10 mol%), base, oxidant, solvent (1.5 mL) in an open flask at 60 °C for 2 h. b Unless otherwise specified, NH3·H2O (25%) was 0.25 mL, other bases were 0.4 mmol. c Yields were determined by 1H NMR. d At 70 °C. e At room temperature. f 2a (0.4 mmol). | |||||
| 1 | CuSO4·5H2O | NH3·H2O | MeCN | Air | 43 |
| 2 | CuBr | NH3·H2O | MeCN | Air | 14 |
| 3 | CuBr2 | NH3·H2O | MeCN | Air | 43 |
| 4 | Cu2O | NH3·H2O | MeCN | Air | 25 |
| 5 | CuO | NH3·H2O | MeCN | Air | 37 |
| 6 | CuI | NH3·H2O | MeCN | Air | Trace |
| 8 | CuCl | NH3·H2O | MeCN | Air | Trace |
| 9 | Cu(OTf)2 | NH3·H2O | MeCN | Air | 40 |
| 10 | CuSO4·5H2O | — | MeCN | Air | 26 |
| 11 | CuSO4·5H2O | Cs2CO3 | MeCN | Air | 25 |
| 12 | CuSO4·5H2O | K2CO3 | MeCN | Air | 33 |
| 13 | CuSO4·5H2O | NaOAc | MeCN | Air | 33 |
| 14 | CuBr2 | (iPr)2NEt | MeCN | Air | Trace |
| 15 | CuBr2 | Et3N | MeCN | Air | 18 |
| 16 | CuBr2 | Pyridine | MeCN | Air | 6 |
| 17 | CuSO4·5H2O | NH3·H2O | MeCN | H2O2 | 12 |
| 18 | CuSO4·5H2O | NH3·H2O | MeCN | TBHP | 90 |
| 19 | CuSO4·5H2O | NH3·H2O | MeCN | BQ | 16 |
| 20 | CuSO4·5H2O | NH3·H2O | MeCN | K2S2O8 | 48 |
| 21 | CuSO4·5H2O | NH3·H2O | MeCN | DTBP | Trace |
| 22 | CuSO4·5H2O | NH3·H2O | MeCN | O2 | 30 |
| 23 | CuSO4·5H2O | NH3·H2O | DMF | TBHP | 24 |
| 24 | CuSO4·5H2O | NH3·H2O | DMSO | TBHP | 56 |
| 25 | CuSO4·5H2O | NH3·H2O | EtOH | TBHP | 52 |
| 26 | CuSO4·5H2O | NH3·H2O | 1,4-Dioxane | TBHP | 28 |
| 27 | — | NH3·H2O | MeCN | TBHP | 0 |
| 28 | CuSO4·5H2O | NH3·H2O | MeCN | TBHP | 80d(5)e |
| 29f | CuSO4·5H2O | NH3·H2O | MeCN | TBHP | 70 |
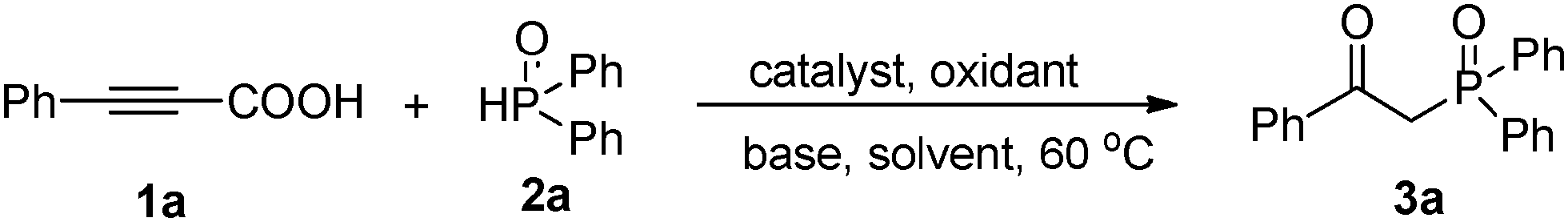
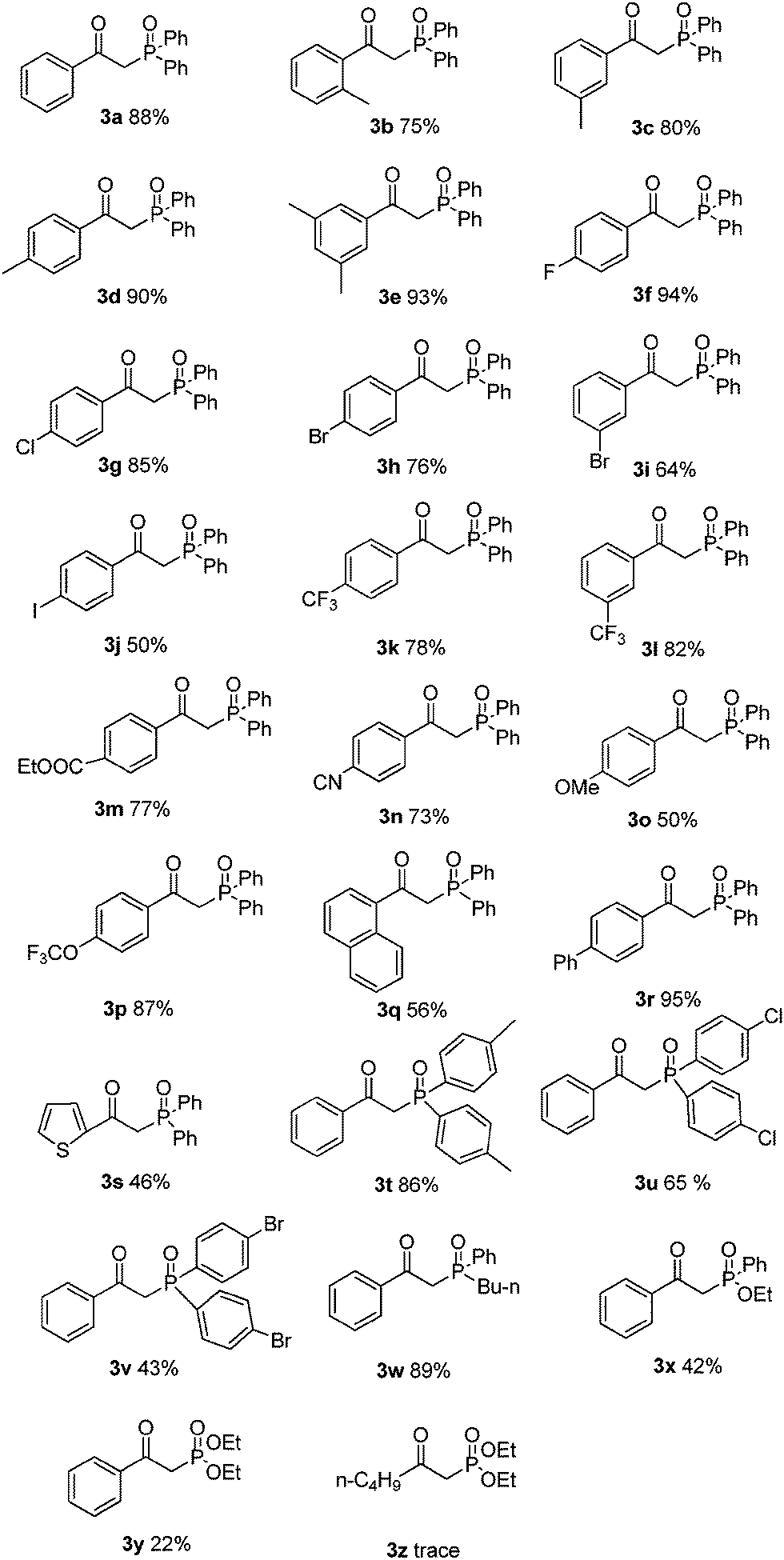
With this preliminary result in hand, the generality of the method was explored under the optimized conditions [alkynyl acid (0.2 mmol), P(O)–H (0.8 mmol), CuSO4·5H2O (10 mmol%), TBHP (2 equiv.), NH3·H2O (25%, 0.25 mL) in MeCN at 60 °C in air for 2 h], and the results are summarized in Table 2. In general, a variety of functional groups on the phenyl ring of arylpropiolic acids were compatible using this procedure, affording the desired products in good to excellent yields. The methyl substituted arylpropiolic acids, such as meta-methyl, para-methyl, and 3,5-dimethyl groups on the aryl ring, reacted with 2a efficiently and gave the desired products 3c–3e in high yields. The ortho-substituted arylpropiolic acids exhibited a particularly distinct steric hindrance effect (3b–3e), and the corresponding β-ketophosphine oxide 3b was obtained in a slightly lower yield (75%). Halogen atoms such as fluoro, chloro, bromo, and iodo on the aromatic ring were unaffected under the present reaction conditions to afford the corresponding products 3f–3j in good yields, which could allow for further synthetic transformations. Arylpropiolic acids bearing electron-withdrawing CF3, COOEt, CN groups reacted smoothly to give the corresponding products in good yields (3k–3n). Treatment of diphosphine oxide with methoxy-substituted arylpropiolic acid led to the formation of product 3o in 50% yield. Replacing the methoxy group with the trifluoromethoxy group resulted in a higher yield (3p, 87%). More bulky substrates such as 3-(naphthalen-1-yl)propiolic acid also smoothly reacted with diphosphine oxide and gave product 3q in 56% yield. In addition, 3-(thiophen-2-yl)propiolic acid could also provide the expected product 3s in 46% yield.
|
|
|---|
|
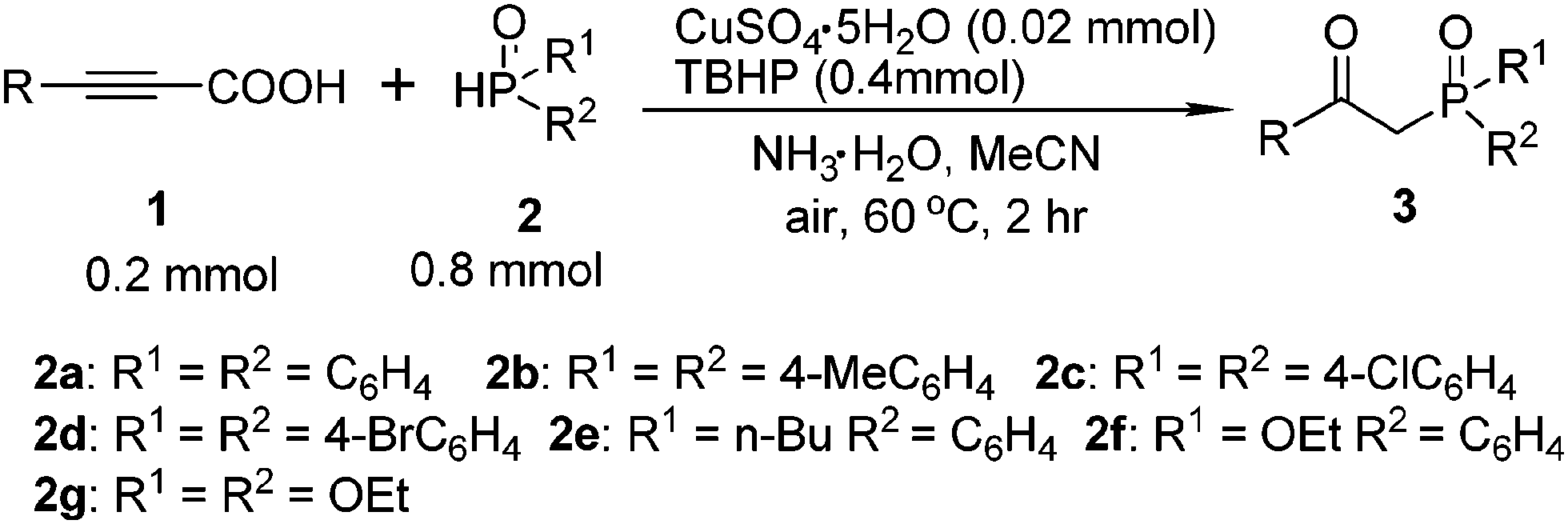
The substrate scope was further investigated by reacting phenylpropiolic acid (1a) with different organophosphorous reagents. Apart from 2a, di-p-tolylphosphine oxide (2b) and bis(4-chlorophenyl)phosphine oxide (2c) were all suitable substrates, generating the corresponding products 3t and 3u in 86% and 65% yields, respectively. However, diarylphosphine oxide involving a para-bromo substituent 2d produced the desired product 3v in only 43% yield. The butyl(p-tolyl)phosphine oxide (2e) also efficiently reacted with 1a and led to the corresponding product 3w in 89% yield. Treatment of ethyl phenylphosphinate (2f) with 1a afforded the desired product 3x in a lower yield of 42%. When diethyl phosphonate (2g) was used, β-ketophosphonate 3y was obtained in only 22% yield. Alkynyl acid was also examined. Unfortunately, only a trace amount of the desired product was detected by 31P NMR analysis.
With the synthetic β-ketophosphine oxides in hand, we next prepared α-benzyl β-ketodiphenylphosphine oxide 4 from benzyl bromide and 3a in good yield, which was converted into 1,3-diphenylpropan-1-one 5 in 98% yield via a dephosphinoylation process (Scheme 2).
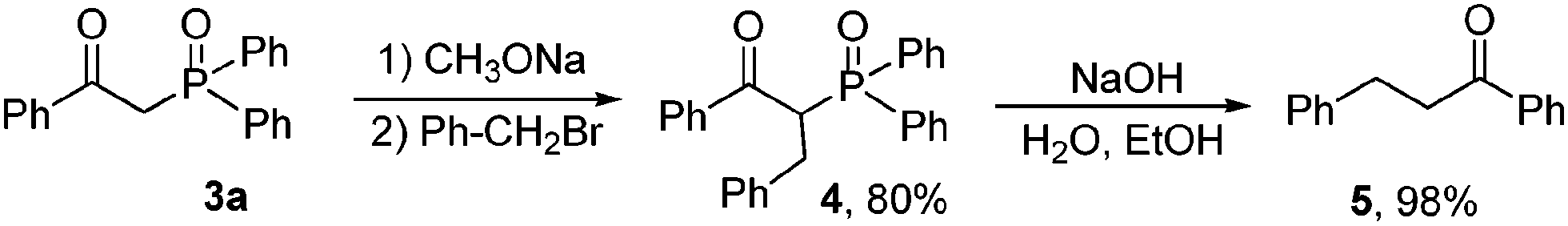 | ||
| Scheme 2 Synthesis of 1,3-diphenylpropan-1-one. | ||
In an effort to improve our understanding of the reaction profile, a series of isotope labeling studies were conducted. When H218O or D2O was added to the reaction mixture under the optimal conditions, no isotope-labeled product was detected in 1H NMR and ESI-MS spectra (Scheme 3a and b). In the absence of air, the transformation could still proceed smoothly to provide 3a in a good yield of 78% (Scheme 3c). It was suggested that the oxygen of the newly formed carbonyl group of 3a mainly originated from TBHP. When 2a was treated with 1.2 equiv. of (phenylethynyl)copper under the optimized reaction conditions, only a trace amount of the desired product was observed, illustrating that (phenylethynyl)copper might not be an intermediate in this process (d). A radical scavenger such as TEMPO could completely restrain the reaction, thus suggesting that the radical processes might be involved. Based on these experimental results and previous mechanistic studies, a plausible mechanism is proposed as shown in Scheme 4.
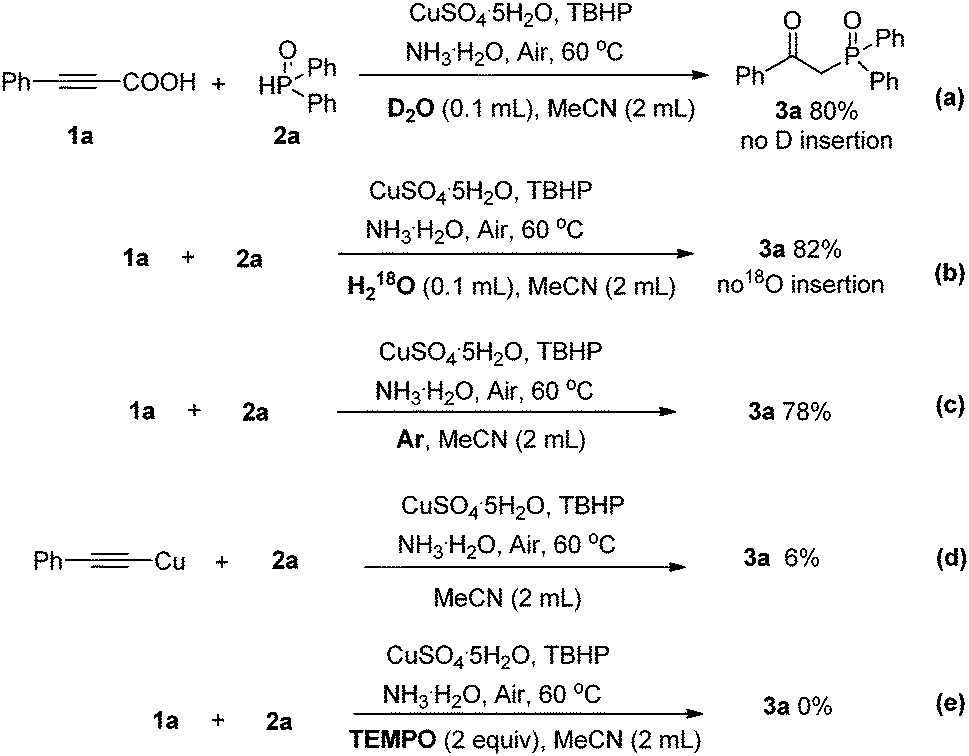 | ||
| Scheme 3 Experiments for the mechanistic study. | ||
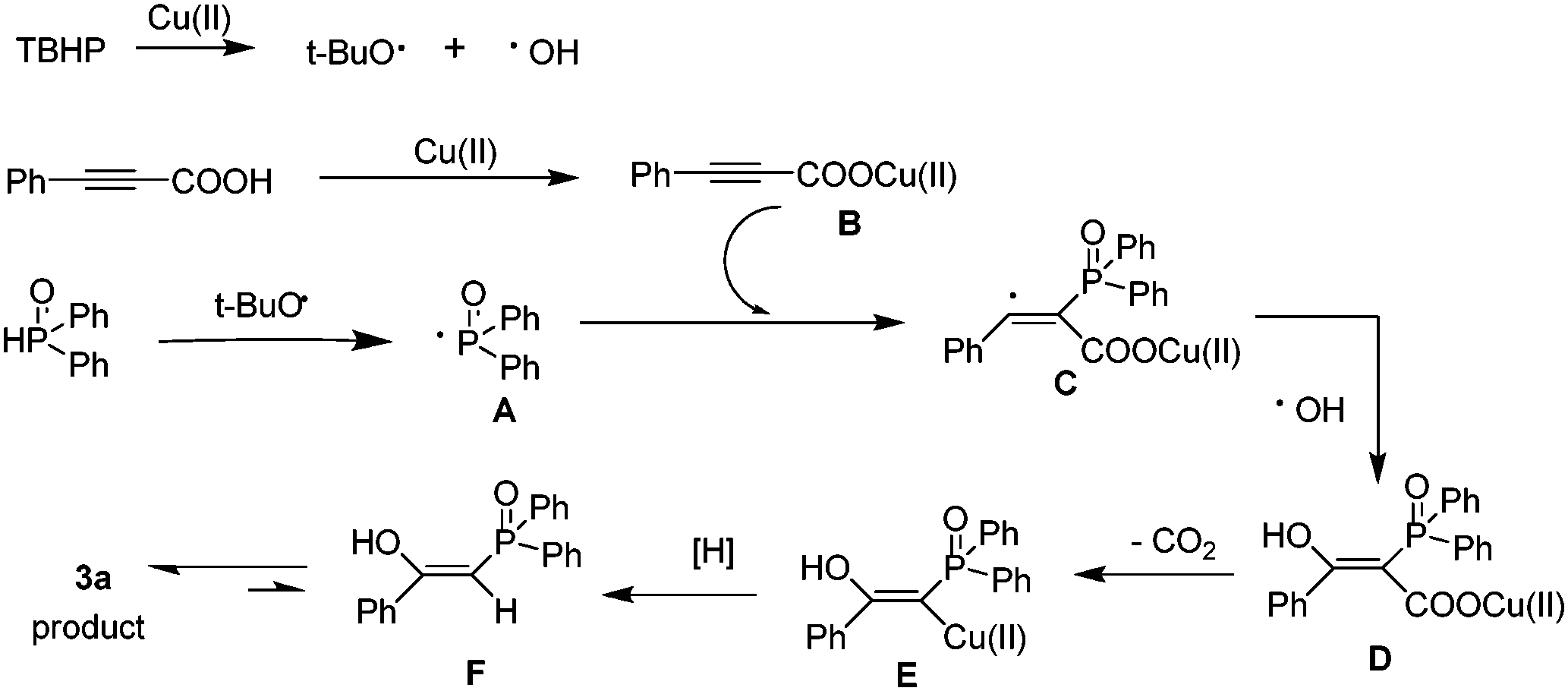 | ||
| Scheme 4 Proposed reaction mechanism. | ||
First, TBHP generates the tert-butoxy radical and the hydroxyl radical in the presence of Cu(II). Then, the tert-butoxy radical triggers the formation of phosphorus radical A from H(O)PPh2. Reaction of the phenylpropiolic acid with Cu(II) generates a salt of cupric carboxylate B. Subsequently, addition of phosphorus radical A at the α-position of the triple bond of B gives intermediate C, which is ultimately trapped by the hydroxyl radical to form intermediate D. Then D releases one molecular CO2 to produce alkenyl copper intermediate E. Finally, the protonolysis of the intermediate leads to the formation of F, which isomerizes and affords the desired product.
In conclusion, we have successfully developed the first facile method for the preparation of β-ketophosphine oxides via decarboxylation–phosphination–oxidation of various alkynyl acids with H-phosphine oxides. Importantly, this transformation would provide a new pathway for formation of Csp3–P and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in one step. This method is highly efficient and provides a rapid access to a broad spectrum of β-ketophosphine oxides in good to excellent yields. Moreover, the use of an inexpensive CuSO4·5H2O catalyst, using readily available alkynyl acids only producing CO2 means that this facile protocol will be attractive for academia and industry.
O bonds in one step. This method is highly efficient and provides a rapid access to a broad spectrum of β-ketophosphine oxides in good to excellent yields. Moreover, the use of an inexpensive CuSO4·5H2O catalyst, using readily available alkynyl acids only producing CO2 means that this facile protocol will be attractive for academia and industry.
We acknowledge financial support from the Chinese National Natural Science Foundation (21173178, 21232005, and 21375113), GD2012-D01-001, the National Basic Research Program of China (2012CB821600), and the Program for Changjiang Scholars and Innovative Research Team in University.
Notes and references
- (a) T. Imamoto, S.-i. Kikuchi, T. Miura and Y. Wada, Org. Lett., 2001, 3, 87 CrossRef CAS; (b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681 CrossRef CAS PubMed; (c) A. R. Choudhury and S. Mukherjee, Adv. Synth. Catal., 2013, 355, 1989 CrossRef CAS; (d) X.-T. Ma, Y. Wang, R.-H. Dai, C.-R. Liu and S.-K. Tian, J. Org. Chem., 2013, 78, 11071 CrossRef CAS PubMed.
- Selected publications: (a) H. H. Chou and C. H. Cheng, Adv. Mater., 2010, 22, 2468 CrossRef CAS PubMed; (b) V. Spampinato, N. Tuccitto, S. Quici, V. Calabrese, G. Marletta, A. Torrisi and A. Licciardello, Langmuir, 2010, 26, 8400 CrossRef CAS PubMed; (c) S. Monge, B. Canniccioni, A. Graillot and J. J. Robin, Biomacromolecules, 2011, 12, 1973 CrossRef CAS PubMed; (d) Y. J. Cho and J. Y. Lee, Chem. – Eur. J., 2011, 17, 11415 CrossRef CAS PubMed; (e) S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 7239 RSC.
- Some selected publications: (a) T. S. Kumar, S. Y. Zhou, B. V. Joshi, R. Balasubramanian, T. H. Yang, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2010, 53, 2562 CrossRef CAS PubMed; (b) F. R. Alexandre, A. Amador, S. Bot, C. Caillet, T. Convard, J. Jakubik, C. Musiu, B. Poddesu, L. Vargiu, M. Liuzzi, A. Roland, M. Seifer, D. Standring, R. Storer and C. B. Dousson, J. Med. Chem., 2011, 54, 392 CrossRef CAS PubMed; (c) X. Chen, D. J. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu, C. Guimaraes, S. Shen, S. Li, S. Johnstone, S. Thibault, H. Xu, M. Cardozo, W. Shen, N. Walker, F. Kayser and Z. Wang, J. Med. Chem., 2012, 55, 3837 CrossRef CAS PubMed.
- (a) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151 CrossRef CAS PubMed; (b) J. Fischer, M. Schürmann, M. Mehring, U. Zachwieja and K. Jurkschat, Organometallics, 2006, 25, 2886 CrossRef CAS; (c) M. N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC.
- J. Clayden and S. Warren, Angew. Chem., Int. Ed. Engl., 1996, 35, 241 CrossRef CAS.
- T. Boesen, D. J. Fox, W. Galloway, D. S. Pedersen, C. R. Tyzack and S. Warren, Org. Biomol. Chem., 2005, 3, 630 CAS.
- D. J. Fox, D. S. Pedersen and S. Warren, Org. Biomol. Chem., 2006, 4, 3102 CAS.
- (a) C. N. Lestas and M. R. Truter, J. Chem. Soc. A, 1971, 738 RSC; (b) D. J. McCabe, E. N. Duesler and R. T. Paine, Inorg. Chem., 1985, 24, 4626 CrossRef CAS.
- R. S. Torr and S. Warren, J. Chem. Soc. Pak., 1979, 1, 15 CAS.
- (a) X. Li, G. Hu, P. Luo, G. Tang, Y. Gao, P. Xu and Y. Zhao, Adv. Synth. Catal., 2012, 354, 2427 CrossRef CAS; (b) X. Chen, X. Li, X.-L. Chen, L.-B. Qu, J.-Y. Chen, K. Sun, Z.-D. Liu, W.-Z. Bi, Y.-Y. Xia, H.-T. Wu and Y.-F. Zhao, Chem. Commun., 2015, 51, 3846 RSC.
- (a) F. Orsini, E. D. Teodoro and M. Ferrari, Synthesis, 2002, 1683 CrossRef CAS; (b) F. Orsini and A. Caselli, Tetrahedron Lett., 2002, 43, 7259 CrossRef CAS; (c) L. Xie, R. Yuan, R. Wang, Z. Peng, J. Xiang and W. He, Eur. J. Org. Chem., 2014, 2668 CrossRef CAS; (d) W. Wei and J. X. Li, Angew. Chem., Int. Ed., 2011, 50, 9097 ( Angew. Chem. , 2011 , 123 , 9263 ) CrossRef CAS PubMed; (e) G. P. Luke, C. K. Seekamp, Z.-Q. Wang and B. L. Chenard, J. Org. Chem., 2008, 73, 6397 CrossRef CAS PubMed.
- M. Nilsson, Acta Chem. Scand., 1966, 20, 423 CrossRef CAS PubMed.
- Selected recent reviews: (a) K. Park and S. Lee, RSC Adv., 2013, 3, 14165 RSC; (b) N. Rodríguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (c) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed . Selected recent papers: ; (d) L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef CAS PubMed; (e) F. Yin, Z. T. Wang, Z. D. Li and C. Z. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (f) J. Hu, N. Zhao, B. Yang, G. Wang, L.-N. Guo, Y.-M. Liang and S.-D. Yang, Chem. – Eur. J., 2011, 17, 5516 CrossRef CAS PubMed; (g) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef CAS PubMed; (h) B. Song, T. Knauber and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2954 CrossRef CAS PubMed.
- X. Li, F. Yang, Y. Wu and Y. Wu, Org. Lett., 2014, 16, 992 CrossRef CAS PubMed.
- G. Hu, Y. Gao and Y. Zhao, Org. Lett., 2014, 16, 4464 CrossRef CAS PubMed.
- (a) S. Van der Jeught and C. V. Stevens, Chem. Rev., 2009, 109, 2672 CrossRef CAS PubMed; (b) M. Mondal and U. Bora, RSC Adv., 2013, 3, 18716 RSC; (c) Y. Gao, J. Wu, J. Xu, X. Wang, G. Tang and Y. Zhao, Asian J. Org. Chem., 2014, 3, 691 CrossRef CAS; (d) Y. M. Li, M. Sun, H. L. Wang, Q. P. Tian and S. D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed; (e) B. Zhang, C. G. Daniliuc and A. Studer, Org. Lett., 2014, 16, 250 CrossRef CAS PubMed; (f) H. C. Fisher, O. Berger, F. Gelat and J. L. Montchamp, Adv. Synth. Catal., 2014, 356, 1199 CrossRef CAS; (g) D. P. Li, X. Q. Pan, L. T. An, J. P. Zou and W. Zhang, J. Org. Chem., 2014, 79, 1850 CrossRef CAS PubMed; (h) Z. Zhao, W. Xue, Y. Gao, G. Tang and Y. Zhao, Chem. – Asian J., 2013, 8, 713 CrossRef CAS PubMed.
- (a) J. Xu, P. Zhang, Y. Gao, Y. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2013, 78, 8176 CrossRef CAS PubMed; (b) Y. Gao, Z. Huang, R. Zhuang, J. Xu, P. Zhang, G. Tang and Y. Zhao, Org. Lett., 2013, 15, 4214 CrossRef CAS PubMed.
- (a) Y. Gao, J. Wu, J. Xu, P. Zhang, G. Tang and Y. Zhao, RSC Adv., 2014, 4, 51776 RSC; (b) Y. Gao, X. Li, J. Xu, Y. Wu, W. Chen, G. Tang and Y. Zhao, Chem. Commun., 2015, 51, 1605 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis, spectral data and NMR spectra of compounds 3a–3z. See DOI: 10.1039/c5cc01904d |
| This journal is © The Royal Society of Chemistry 2015 |