
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inverse regioselectivity in the silylstannylation of alkynes and allenes: copper-catalyzed three-component coupling with a silylborane and a tin alkoxide†
H.
Yoshida
*ab,
Y.
Hayashi
a,
Y.
Ito
a and
K.
Takaki
a
aDepartment of Applied Chemistry, Graduate School of Engineering, Hiroshima University, Higashi-Hiroshima 739-8527, Japan. E-mail: yhiroto@hiroshima-u.ac.jp; Fax: +81-82-424-5494; Tel: +81-82-424-7724
bACT-C, Japan Science and Technology Agency, Higashi-Hiroshima 739-8527, Japan
First published on 14th April 2015
Abstract
Silylstannylation of alkynes and allenes has been found to proceed by three-component coupling using a silylborane and a tin alkoxide in the presence of a Cu(I) catalyst. The regioselectivities are completely inverse to those of the conventional silylstannylation under palladium catalysis.
Transition metal-catalyzed dual functionalization of a carbon–carbon multiple bond of unsaturated hydrocarbons such as alkynes and allenes with metallic elements (dimetallation)1 has attracted considerable attention as a convenient and straightforward entry to synthetically potent vic-dimetallated compounds of defined structure, whose carbon–metal bonds are utilizable for construction of carbon frameworks2 and introduction of functional groups. One of the most prevailing dimetallations is silylstannylation,3 which has thus far been shown to proceed through direct addition of a silicon–tin bond of silylstannanes across unsaturated carbon linkages under palladium catalysis. The characteristic feature of the palladium-catalyzed silylstannylation is a high level of regioselectivity: the silyl addition commonly occurs at the terminal carbon of terminal alkynes3a–f and at the central carbon of allenes,3g–j irrespective of electronic and steric characters of ligands employed and substituents on the carbon–carbon multiple bonds (Scheme 1).4
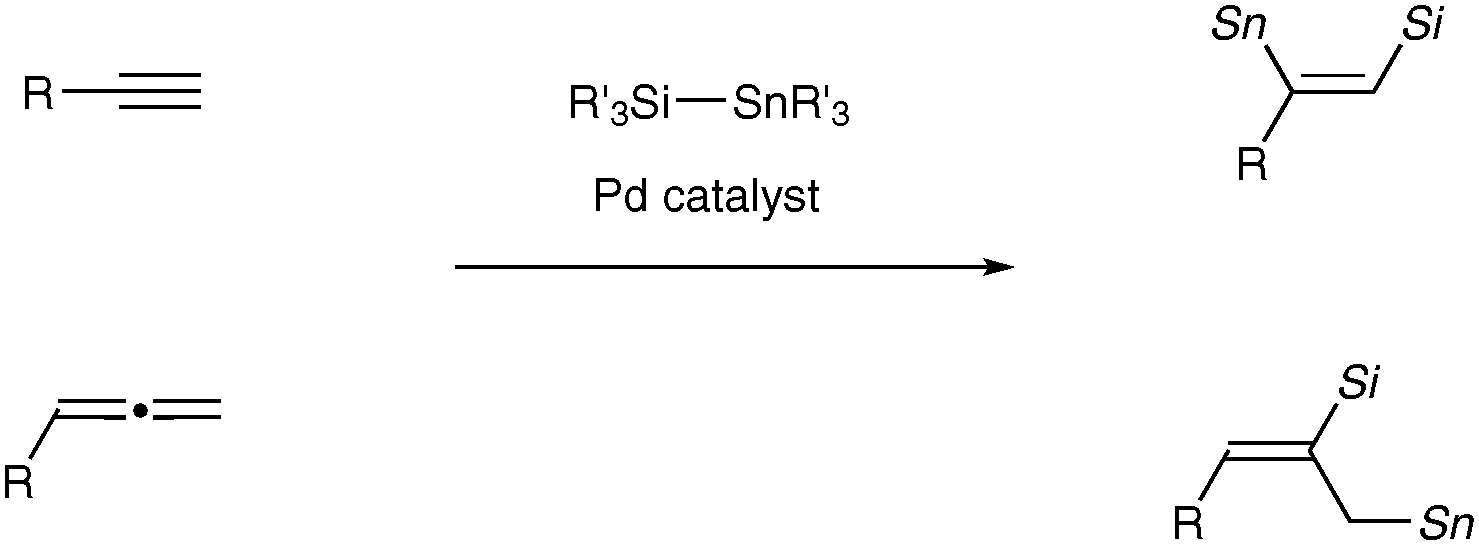 | ||
| Scheme 1 Regioselectivity in Pd-catalyzed silylstannylation. | ||
Recently, we have devoted our attention to exploitation of potential copper catalysis toward the dimetallation of unsaturated hydrocarbons, and have already disclosed that diborylation,5a distannylation5b and three-component borylstannylation5c–e of carbon–carbon multiple bonds take place in an unique reaction mode,6 where the oxidation state of a copper catalyst stays constant throughout the reaction, being in marked contrast to the conventional oxidative addition–insertion–reductive elimination sequence, which generally involves two-electron redox of a transition metal catalyst.1,7 The key intermediates in these transformations are β-boryl (or stannyl)organocopper species arising from insertion of unsaturated hydrocarbons into boryl (or stannyl)copper species, and capturing them by a tin alkoxide finally affords the respective stannylated products as depicted in Scheme 2.5b–e Thus, we envisaged that silylstannylation of unsaturated hydrocarbons would also be feasible under copper catalysis by use of a suitable silylating reagent which allows facile generation of silylcopper8 and β-silylorganocopper species.
 | ||
| Scheme 2 Cu-catalyzed borylstannylation and distannylation. | ||
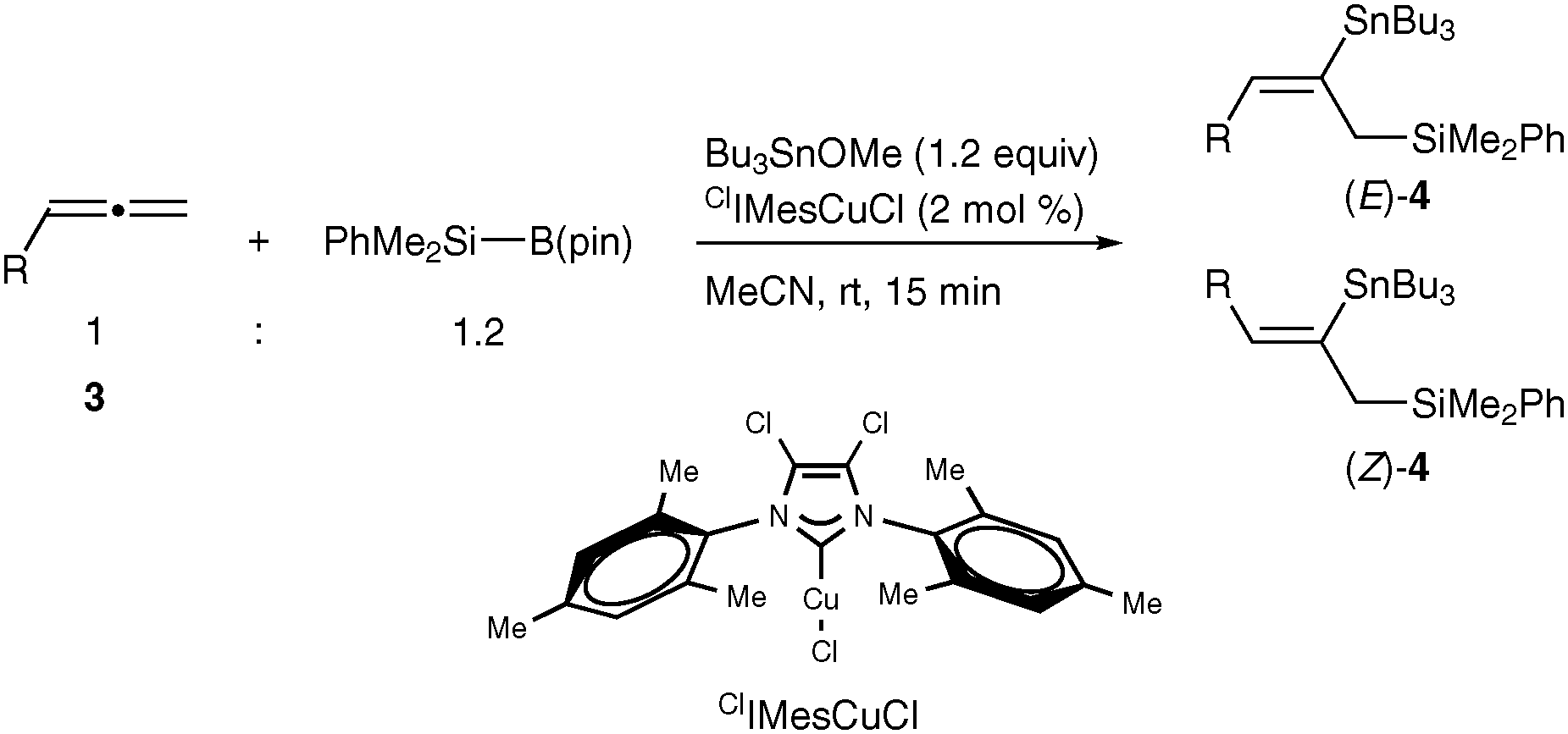
Herein we report that the silylstannylation of alkynes and allenes facilely occurs by the copper-catalyzed three-component coupling with a silylborane,9 and that the regioselectivities become totally inverse to those of the conventional silylstannylation3 in both cases.
The three-component silylstannylation was found to readily occur to afford 2a and 2′a in 86% yield with regioselectivity inverse to those of the previous Pd-catalyzed silylstannylation (2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2′a = 93:7), when the reaction of 1-octyne (1a), a silylborane (PhMe2Si–B(pin), pin: pinacolato) and tributyltin tert-butoxide10 was carried out in acetonitrile at room temperature in the presence of a CuCl–P(tBu)3 catalyst (Table 1, entry 1). Although the silylstannylation products were also formed with other monodentate phosphines (PPh3, JohnPhos and Cy-JohnPhos) and N-heterocyclic carbene (IMes), the yields and the regioselectivities were unsatisfactory (entries 2–5). Acetonitrile has been proven to be the solvent of choice: the reaction in less polar solvents (toluene and THF) produced a hydrosilylation product, nHex(PhMe2Si)C
:2′a = 93:7), when the reaction of 1-octyne (1a), a silylborane (PhMe2Si–B(pin), pin: pinacolato) and tributyltin tert-butoxide10 was carried out in acetonitrile at room temperature in the presence of a CuCl–P(tBu)3 catalyst (Table 1, entry 1). Although the silylstannylation products were also formed with other monodentate phosphines (PPh3, JohnPhos and Cy-JohnPhos) and N-heterocyclic carbene (IMes), the yields and the regioselectivities were unsatisfactory (entries 2–5). Acetonitrile has been proven to be the solvent of choice: the reaction in less polar solvents (toluene and THF) produced a hydrosilylation product, nHex(PhMe2Si)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, as a by-product (entries 6 and 7), and the regioselectivity became lower with DMF (entry 8).
CH2, as a by-product (entries 6 and 7), and the regioselectivity became lower with DMF (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Time (h) | Yieldb (%) |
2a:2′a |
|
a General procedure: 1a (0.30 mmol), PhMe2Si–B(pin) (0.36 mmol), Bu3SnOtBu (0.36 mmol), CuCl (6.0 μmol), ligand (6.0 μmol), solvent (1 mL).
b Isolated yield.
c NMR yield.
d IMesCuCl (2 mol%) was used.
e A hydrosilylation product, nHex(PhMe2Si)CCH2, was formed in 6% NMR yield.
f A hydrosilylation product, nHex(PhMe2Si)CCH2, was formed in 10% NMR yield.
|
|||||
| 1 | P(tBu)3 | MeCN | 6 | 86 | 93:7 |
| 2 | PPh3 | MeCN | 3 | 88c | 80:20 |
| 3 | JohnPhos | MeCN | 23 | 58c | 86:14 |
| 4 | Cy-JohnPhos | MeCN | 27 | 54c | 85:15 |
| 5 | IMesd | MeCN | 3 | 84c | 75:25 |
| 6e | P(tBu)3 | Toluene | 4 | 69 | 99:1 |
| 7f | P(tBu)3 | THF | 3 | 63 | 99:1 |
| 8 | P(tBu)3 | DMF | 1.5 | 79 | 90:10 |

Under the optimized reaction conditions, 1-hexyne (1b), 1-decyne (1c) and branched aliphatic terminal alkynes (1d–1f) could undergo the regioselective silylstannylation, where the stannyl moieties were predominantly attached to the terminal carbon of the alkynes (Table 2, entries 1–5). This unique regioselectivity was also achievable with functionalized alkynes bearing a cyano (1g), bromo (1h), hydroxy (1i) or amino (1j) group (entries 6–9), and the results that these reactive moieties remained intact demonstrate the high functional group compatibility of the silylstannylation. In contrast, the stannyl moiety was selectively introduced into the internal carbon of THP-protected propargyl alcohol (1k) and propargyl ether (1l) to provide 2′k and 2′l as the major products (entries 10 and 11), and the reaction of enyne (1m) or phenylacetylene (1n) resulted in low regioselectivity (entries 12 and 13).
|
|
||||
|---|---|---|---|---|
| Entry | R | Time (h) | Yieldb (%) |
2:2′ |
| a General procedure: 1 (0.30 mmol), PhMe2Si–B(pin) (0.36 mmol), Bu3SnOtBu (0.36 mmol), CuCl (6.0 μmol), P(tBu)3 (6.0 μmol), MeCN (1 mL). b Isolated yield. c Cyp = cyclopentyl. | ||||
| 1 | nBu (1b) | 3 | 76 | 95:5 |
| 2 | nOct (1c) | 1.5 | 68 | 94:6 |
| 3 | Cypc (1d) | 7 | 65 | 93:7 |
| 4 | iBu (1e) | 4 | 64 | 97:3 |
| 5 | iAmyl (1f) | 19 | 61 | 90:10 |
| 6 | NC(CH2)3 (1g) | 3.5 | 56 | 94:6 |
| 7 | Br(CH2)2 (1h) | 16 | 47 | 91:9 |
| 8 | HO(CH2)2 (1i) | 4 | 43 | 97:3 |
| 9 | Et2NCH2 (1j) | 25.5 | 39 | 90:10 |
| 10 | THPOCH2 (1k) | 6.5 | 75 | 10:90 |
| 11 | MeOCH2 (1l) | 12.5 | 59 | 1:99 |
| 12 | 1-Cyclohexenyl (1m) | 27 | 61 | 61:39 |
| 13 | Ph (1n) | 7 | 64 | 14:86 |

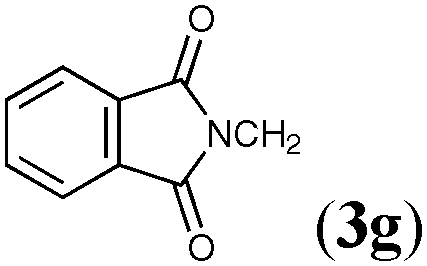
The three-component silylstannylation of allenes was found to also proceed smoothly with regioselectivity inverse to those of the previous silylstannylation under palladium catalysis. Thus, treatment of pentadeca-1,2-diene (3a) with a silylborane and tributyltin methoxide11 in the presence of the ClIMesCuCl catalyst12 afforded an 87% yield of (E)- and (Z)-4a (ratio = 78:22), whose stannyl moiety was exclusively installed into the central carbon of the allene (Table 3, entry 1). The regioselective formation of silylstannylated products (4b–4d) bearing allylsilane and alkenylstannane units was observed with 5-phenyl-penta-1,2-diene (3b), cyclohexylallene (3c) and undeca-1,2-diene (3d) (entries 2–4), and furthermore functionalized allenes possessing a silyl ether (3e), a theobromine (3f), a phthalimide (3g) or an acetal (3h) moiety underwent the silylstannylation with a similar regioselectivity to provide the respective products (4e–4h) without damaging these functional groups (entries 5–8).
|
|
|||
|---|---|---|---|
| Entry | R | Yieldb (%) |
E:Z |
| a General procedure: 3 (0.30 mmol), PhMe2Si–B(pin) (0.36 mmol), Bu3SnOMe (0.36 mmol), ClIMesCuCl (6.0 μmol), MeCN (1 mL). b Isolated yield. | |||
| 1 | Dodecyl (3a) | 87 | 78:22 |
| 2 | Ph(CH2)2 (3b) | 86 | 77:23 |
| 3 | Cy (3c) | 77 | 87:13 |
| 4 | nOct (3d) | 71 | 91:9 |
| 5 | TBSO(CH2)2 (3e) | 93 | 75:25 |
| 6 |

|
76 | 83:17 |
| 7 |
|
63 | 79:21 |
| 8 | THPOCH2 (3h) | 56 | 75:25 |
Generation of a silylcopper species, Cu–SiMe2Ph, via σ-bond metathesis between a copper alkoxide and a silylborane would trigger the silylstannylation (Scheme 3, step A).13 Then an alkyne or an allene was inserted into the Cu–Si bond to give a β-silylalkenylcopper species (5 or 6) (step B),14 which was subsequently trapped by a tin alkoxide to furnish a silylstannylation product with regeneration of a copper alkoxide (step C).15–17 The regiochemical outcome of the reaction with an alkyne or an allene should be ascribable to the regioselective formation of 518 or 6, the latter of which has been demonstrated to be kinetically favored in the stoichiometric reaction using a silylcopper species.8a On the other hand, electronic directing effect of a propargylic functional group (1k and 1l) or a phenyl group (1n), which induces the addition of the copper moiety to the internal carbon of the alkynes in step B,19 should become dominant to provide 2′ as the major product.
 | ||
| Scheme 3 A plausible catalytic cycle for silylstannylation. | ||
In conclusion, we have demonstrated that the regioselectivities of the silylstannylation of terminal alkynes and allenes can totally be reversed depending upon the copper-catalyzed three-component coupling using a silylborane and a tin alkoxide, which leads to convenient and direct access to diverse 2-silyl-1-stannyl-1-alkenes (from alkynes) and 1-silyl-2-stannyl-2-alkenes (from allenes) of high synthetic utility. Further studies on copper-catalyzed silylation reactions of unsaturated carbon–carbon bonds as well as synthetic application of the silylstannylation are in progress.
Notes and references
- For reviews, see: (a) I. Beletskaya and C. Moberg, Chem. Rev., 1999, 99, 3435 CrossRef CAS PubMed; (b) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221 CrossRef CAS PubMed; (c) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed.
- Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VHC, Weinheim, 2004 Search PubMed.
- For representative silylstannylation of alkynes, see: (a) B. L. Chenard, E. D. Laganis, F. Davidson and T. V. RajanBabu, J. Org. Chem., 1985, 50, 3666 CrossRef CAS; (b) T. N. Mitchell, H. Killing, R. Dicke and R. Wickenkamp, J. Chem. Soc., Chem. Commun., 1985, 354 RSC; (c) T. N. Mitchell, R. Wickenkamp, A. Amamria, R. Dicke and U. Schneider, J. Org. Chem., 1987, 52, 4868 CrossRef CAS; (d) I. Hemeon and R. D. Singer, Chem. Commun., 2002, 1884 RSC; (e) M. Murakami, T. Matsuda, K. Itami, S. Ashida and M. Terayama, Synthesis, 2004, 1522 CrossRef CAS PubMed; (f) T. E. Nielsen, S. Le Quement and D. Tanner, Synthesis, 2004, 1381 CAS ; For representative silylstannylation of allenes, see: ; (g) T. N. Mitchell and U. Schneider, J. Organomet. Chem., 1991, 407, 319 CrossRef CAS; (h) A. G. M. Barrett and P. W. H. Wan, J. Org. Chem., 1996, 61, 8667 CrossRef CAS; (i) S. Shin and T. V. RajanBabu, J. Am. Chem. Soc., 2001, 123, 8416 CrossRef CAS; (j) M. Jeganmohan, M. Shanmugasundaram, K.-J. Chang and C.-H. Cheng, Chem. Commun., 2002, 2552 RSC.
- An alkoxyalkyne exceptionally accepts the silyl addition at the internal carbon. See: M. Murakami, H. Amii, N. Takizawa and Y. Ito, Organometallics, 1993, 12, 4223 CrossRef CAS.
- (a) H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2012, 51, 235 CrossRef CAS PubMed; (b) H. Yoshida, A. Shinke and K. Takaki, Chem. Commun., 2013, 49, 11671 RSC; (c) Y. Takemoto, H. Yoshida and K. Takaki, Chem. – Eur. J., 2012, 18, 14841 CrossRef CAS PubMed; (d) Y. Takemoto, H. Yoshida and K. Takaki, Synthesis, 2014, 3024 CAS; (e) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2015, 51, 6297 RSC.
- We have also reported other borylation reactions of unsaturated hydrocarbons under copper or silver catalysis. See: (a) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2013, 15, 952 CrossRef CAS PubMed; (b) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2014, 50, 8299 RSC; (c) I. Kageyuki, H. Yoshida and K. Takaki, Synthesis, 2014, 1924 Search PubMed; (d) H. Yoshida, Y. Takemoto and K. Takaki, Asian J. Org. Chem., 2014, 3, 1204 CrossRef CAS PubMed; (e) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2014, 16, 3512 CrossRef CAS PubMed.
- (a) M. Hada, Y. Tanaka, M. Ito, M. Murakami, H. Amii, Y. Ito and H. Nakatsuji, J. Am. Chem. Soc., 1994, 116, 8754 CrossRef CAS; (b) F. Ozawa, Y. Sakamoto, T. Sagawa, R. Tanaka and H. Katayama, Chem. Lett., 1999, 1307 CrossRef CAS; (c) T. Sagawa, Y. Sakamoto, R. Tanaka, H. Katayama and F. Ozawa, Organometallics, 2003, 22, 4433 CrossRef CAS.
- For reviews, see: (a) A. Barbero and F. J. Pulido, Acc. Chem. Res., 2004, 37, 817 CrossRef CAS PubMed; (b) F. J. Pulido and A. Barbero, Silyl and Stannyl Derivatives of Organocopper Compounds, in The Chemistry of Organocopper Compounds Part 2, ed. Z. Rappoport and I. Marek, Wiley, Chichester, 2009, pp. 775–856 Search PubMed.
- For representative examples of copper-catalyzed silylation of unsaturated hydrocarbons with a silylborane, see: (a) P. Wang, X.-L. Yeo and T.-P. Loh, J. Am. Chem. Soc., 2011, 133, 1254 CrossRef CAS PubMed; (b) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487 CrossRef CAS PubMed; (c) F. Meng, H. Jang and A. H. Hoveyda, Chem. – Eur. J., 2013, 19, 3204 CrossRef CAS PubMed; (d) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706 CrossRef CAS PubMed; (e) Y.-H. Xu, L.-H. Wu, J. Wang and T.-P. Loh, Chem. Commun., 2014, 50, 7195 RSC; (f) J. Rae, Y. C. Hu and D. J. Procter, Chem. – Eur. J., 2014, 20, 13143 CrossRef CAS PubMed.
- The reaction with tributyltin methoxide resulted in lower regioselectivity (2a:2′a = 89:11).
- The use of tributyltin tert-butoxide resulted in lower yield. See the ESI† for details.
- For the ligand effect on the silylstannylation of an allene, see the ESI†.
- C. Kleeberg, M. S. Cheung, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 19060 CrossRef CAS PubMed.
- For stoichiometric reactions of a silylcopper species with an alkyne or an allene that produce a β-silylalkenylcopper species, see ref. 8a.
- Generation of a silylcopper species (step A) and insertion of an alkyne or an allene into the Cu–Si bond (step B) have been widely accepted as fundamental elementary steps in the copper-catalyzed silylation reactions of alkynes or allenes with a silylborane. See ref. 9.
- We have already demonstrated that an alkenylcopper species is readily captured with a tin alkoxide to give an alkenylstannane. See ref. 5c.
- Intermediacy of a silylstannane (PhMe2Si–SnBu3) in the present silylstannylation could be ruled out, because the copper-catalyzed reaction of 3d with PhMe2Si–SnBu3 did not produce 4d at all.
- Regioselective formation of this alkenylcopper species was also observed in the copper-catalyzed formal hydrosilylation of terminal alkynes. See ref. 9a.
- A similar regioselectivity was obtained in borylcupration of alkynes. See: (a) H. R. Kim and J. Yun, Chem. Commun., 2011, 47, 2943 RSC; (b) A. L. Moure, R. G. Arrayás, D. J. Cárdenas, I. Alonso and J. C. Carretero, J. Am. Chem. Soc., 2012, 134, 7219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: 10.1039/c5cc01856k |
| This journal is © The Royal Society of Chemistry 2015 |