
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel olfactory ligands via terpene synthases†
Sabrina
Touchet
a,
Keith
Chamberlain
b,
Christine M.
Woodcock
b,
David J.
Miller
a,
Michael A.
Birkett
b,
John A.
Pickett
*b and
Rudolf K.
Allemann
*a
aSchool of Chemistry, Main Building, Cardiff University, Park Place, Cardiff, CF10 3AT, UK. E-mail: allemannrk@cardiff.ac.uk
bBiological Chemistry and Crop Protection Department, Rothamsted Research, Harpenden, Hertfordshire AL5 2JQ, UK. E-mail: john.pickett@rothamsted.ac.uk
First published on 7th April 2015
Abstract
A synthetic biology approach to the rational design of analogues of olfactory ligands by providing unnatural substrates for the enzyme synthesising (S)-germacrene D, an olfactory ligand acting as a plant derived insect repellent, to produce novel ligands is described as a viable alternative to largely unsuccessful ligand docking studies. (S)-14,15-Dimethylgermacrene D shows an unexpected reversal in behavioural activity.
Many organisms employ small lipophilic molecules as external signals for interacting with members of the same species and to locate hosts or food and to avoid antagonistic organisms, e.g. parasites. For animals, the process by which these signals are recognised is olfaction, but the underpinning interactions between the olfactory signal or ligand and the olfactory recognition proteins are similar to those between signals and recognition proteins in organisms from other kingdoms without olfactory nervous systems. Olfactory ligands are, by definition, volatile and may be chemically unstable and expensive to synthesise. Because of their value as key recognition cues in perfumes and cosmetics or food and beverages, and for control of pests, particularly insects, there has been considerable interest in the potential for rational design of analogues with more practically useful and commercially desirable properties. Occasionally, highly active analogues of olfactory ligands1,2 have been found, although attempts to design these have not yielded rational routes, despite the advanced level of understanding of the molecular basis of olfactory ligand recognition. The recognition systems involved are considerably more sophisticated than those recognising ligands acting within organisms, where rational design is now exemplified for in vitro measurements at a range of recognition sites, e.g. for neurotransmitters and hormones, which extend beyond animal systems.3 It is assumed that, by acting externally, olfactory ligands need to be recognised against a far greater diversity of background signals than when the receptor is within an organism and situated beyond externally accessed tissues, e.g. those protected by the blood–brain barrier. This results in a very high specificity for the olfactory recognition process, usually involving true molecular recognition rather than recognition of homologous series of compounds with the same functional group. Thus, for individual compounds, there is a particular difference in smell for structurally related compounds and even the involvement of individual olfactory neurons for specific compounds, as in insect olfaction, that have substantially lower interactions with homologous compounds. Here, we test the hypothesis that, if a compound is accepted as an unnatural substrate by the enzyme that synthesises the natural ligand, then the product is likely to demonstrate sufficient coverage of the chemical space associated with the natural ligand to be active. This proposal was tested using analogues of the sesquiterpene hydrocarbon germacrene D (Fig. 1, (S)-isomer 1; (R)-isomer 2), a highly volatile and unstable olfactory signal that repels invertebrate arthropod pests (insects, ticks, mites) affecting human beings and livestock4 and arable crops,5 and for which the respective synthase enzymes have been characterised previously.6
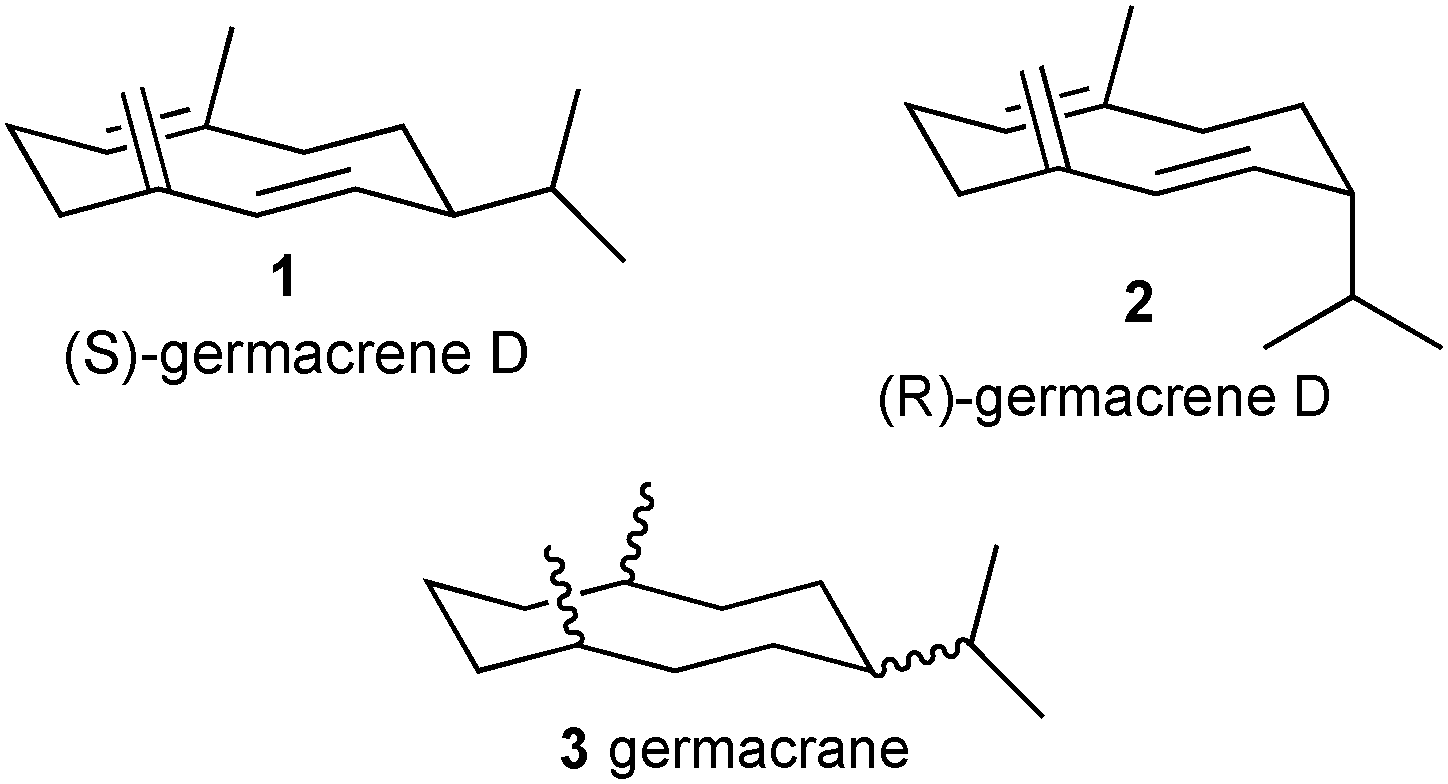 | ||
| Fig. 1 Enantiomers of germacrene D, (S)-germacrene D (1), (R)-germacrene D (2) and germacrane (3). | ||
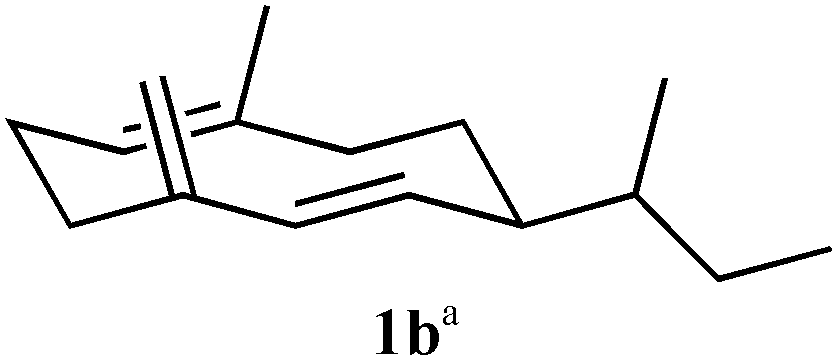
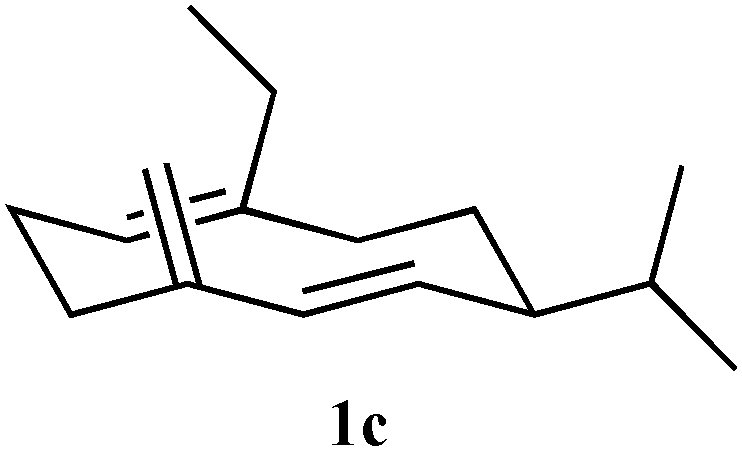
Recombinant (S)-germacrene D synthase containing a C-terminal hexa-histidine tag (GDS) was overproduced in E. coli and purified by Ni2+-affinity chromatography.7 The His-tagged GDS showed similar turnover kinetics to the wild-type enzyme when assayed with [1-3H]-FDP (see ESI†). Unnatural potential substrates, i.e. farnesyl diphosphate (FDP, 4a) analogues7 (Table 1) were synthesised and incubated with GDS under optimised incubation conditions.7,8 Only certain substrate analogues were converted to product (Table 1). The relationship between those converted and those that remained unchanged indicated that certain structural and electronic features were essential for acceptance by GDS. Electron-withdrawing fluoride at either C2 or C10 of the FDP chain led to either little or no substrate activity (for C2) or an alternative 1,11-ring closure to form an α-humulene analogue (5) (for C10).7 Additionally, increased bulk at carbon 15 of FDP led to a loss of substrate activity with less than 1% substrate turnover even after extended (>24 h) incubation times. The enzyme displayed much greater tolerance to changes at C12 and C14; both 12-Me-FDP (4b) and 14-Me-FDP (4c) were good substrates.
|
|
|||||
|---|---|---|---|---|---|
| Substrate | Product | % Conversion | Rel. yieldc | GC-EAG activityf | Olfactometerg |
| 4a R1–R6 = H, also R1–R6 = H for all others except as shown above.a Two products generated, one of which was 1b.b 15F-FDP gave multiple products that decomposed upon standing.c Percentage conversion and percentage conversion relative to natural substrate.4d Isolated yield after preparative scale incubation with GDS-Y406F.e Product was identified as 8-fluoro-α-humulene (5).7f Results from the electroantennograms are expressed as + = active, +++ = highly active.g Results from the olfactometer are expressed as − repellent, − − − highly repellent, +++ highly attractive, n/a = not active, n/t = not tested. | |||||
| 4a (FDP) | 1a | 76 | 100 | +++ | − − − |
| 4b (12Me) R1 = CH3 |
|
46 | 60 | n/a | − − |
| 4c (14Me) R2 = CH3 |
|
49 | 59 | + | n/a |
| 4d (14F) R2 = F |

|
33 | 43 | + | n/t |
| 4e (15F) R3 = F |

|
37 | 49 | n/a | n/t |
| 4f (15Me) R3 = CH3 |

|
<1 | +++ | − − | |
| 45d | |||||
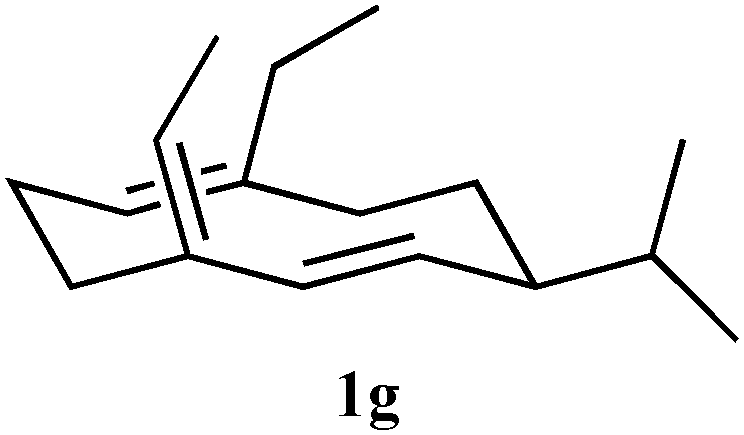
| 4g (14,15di-Me) R2 and R3 = CH3 |
|
<1 | n/a | +++ | |
| 73d | |||||
| 4h (6F) R4 = F |

|
30 | 74 | n/a | n/a |
| 4i (10F) R5 = F, | 5 | <1e | n/t | n/t | |
| 4j (2F) R6 = F | None | − | |||
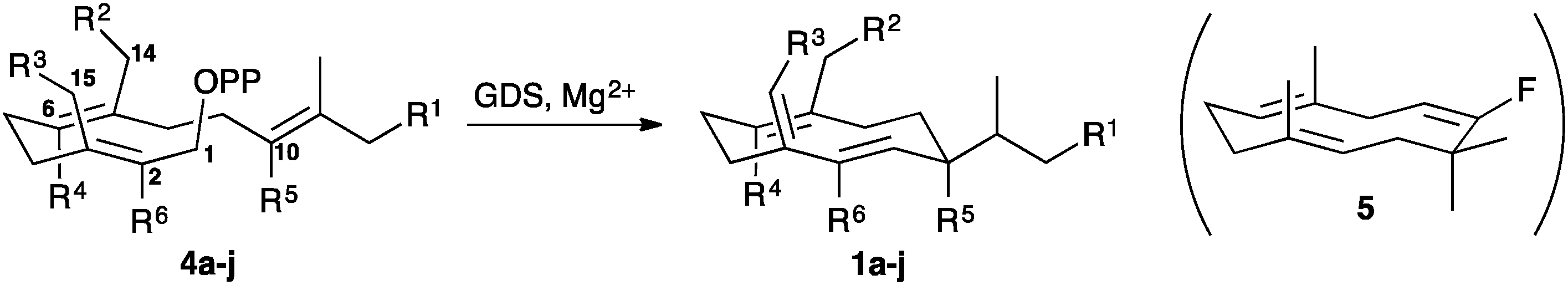
Alterations of amino acid residues both in the active site and in the surrounding regions have previously been shown to modify terpene synthase activity with respect to both product distribution and catalytic efficiency.9–20 To maximise the acceptance of FDP analogues within the active site of GDS, a rational engineering approach was used to tailor the substrate specificity whilst retaining catalytic efficiency and product selectivity. In the absence of a X-ray single crystal structure of GDS, a homology model of GDS was built with I-Tasser21 based on the known structure 5-epi-aristolochene synthase from Nicotiana tabacum coordinated to the inhibitor farnesyl hydroxyphosphonate (FHP) (PDB-5EAT)22 and used to identify aromatic residues that might contribute to cation stabilisation and substrate folding.23 Y524, W275 and Y406 (Fig. 2) were selected to explore the effects of their substitution for other amino acids on GDS catalysis. Hence site directed mutagenesis was performed to replace these residues with amino acids of different sizes and electrostatic and hydrogen-bonding properties. The resulting cDNAs were expressed in E. coli and the corresponding proteins purified as for GDS.7 Each mutant was incubated with FDP (4a), the resulting products were analysed by GC-MS and the reaction kinetics were measured using tritiated FDP (ESI†).
 | ||
| Fig. 2 Homology model of GDS with farnesyl hydroxyphosphonate (FHP) bound at the active site based on the known structure of 5-epi-aristolochene synthase from Nicotiana tabacum coordinated to the inhibitor FHP (PDB-5EAT).22 Active site residues Y406 (blue), Y524 and W275 (orange) that were targeted for site directed mutagenesis are shown along with three Mg2+-cofactor ions (silver spheres). | ||
Replacement of either W275 or Y524 with a variety of residues did not have a positive effect on the catalytic activity. Replacement of Y406 with phenylalanine however did improve the catalytic efficiency of the enzyme. The steady state kinetic parameters of GDS-Y406F revealed a KM 3.5 times higher than that measured for GDS-His6 and a turnover number kcat almost one order of magnitude higher that for the wild-type GDS (ESI†). GDS-Y406F is hence a rare example of a mutant sesquiterpene synthase that is more efficient than its parent (ESI†).
The 14 and 15 positions of FHP (Fig. 2) are close to Y406 of GDS and hence GDS-Y406F should be a more effective enzyme than GDS for the turnover of 15-Me-FDP (4f) and 14,15-diMe-FDP (4g). Incubation of 4f and 4g with GDS-Y406F revealed that the mutant was considerably more effective than wild-type GDS as a synthase for the production of (S)-15-methylgermacrene D (1f) and (S)-14,15-dimethylgermacrene D (1g) (Table 1) with yields of 45% and 73%, respectively. The configuration of the exo-double bond in both compounds was assigned using 1H- and 13C-NMR spectroscopy. The absence of a cross peak in the NOESY spectra (ESI†) between the exo-alkene proton (C15) and the alkene proton at C2 for the germacrene D analogues 1f5 and 1g suggested Z-stereochemistry at the exo-alkene double bond although the absence of a positive correlation means the E isomer cannot be ruled out entirely. Steady state turnover kinetics using tritiated 4a, 4f and 4g (ESI†) showed that relative to wild-type GDS, the catalytic efficiency kcat/KM of the mutant enzyme was approximately 2.4 times higher for each substrate.
To test the hypothesis that the product generated from an unnatural substrate of the enzyme that produces the natural olfactory ligand covers sufficient chemical space to also be an active ligand, electrophysiological and behavioural assays were used on germacrene D analogues produced by GDS and GDS-Y604F. Measurement of electrophysiological activity employed the electroantennogram (EAG) (ESI†), which involves placing electrodes across the antenna of a live aphid, the grain aphid Sitobion avenae, and recording perturbation of the recorded standing potential in comparison with that caused by (S)-germacrene D (both (R)-germacrene D and the fully hydrogenated germacrane (3) were inactive), when the product from the unnatural substrate was presented. Coupled high-resolution gas chromatography-EAG (GC-EAG) was employed to ensure that even minor impurities could not cause misleading effects. Results from GC-EAG and behavioural bioassays using a four-arm olfactometer (ESI†) showed that although a number of the analogues produced by the enzyme were biologically active (Table 1), the activities were not fully in line with the enzyme conversion kinetics. (S)-14-Methylgermacrene D (1c, Table 1) showed activity by GC-EAG but no activity in the olfactometer, and (S)-15-methylgermacrene D (1f, Table 1) was highly active by GC-EAG but was less active than the parent compound in the olfactometer. For (S)-14,15-dimethylgermacrene D (1g, Table 1), the type of repellent behaviours observed with all other electrophysiologically active analogues was reversed, the activity being measured as aphid attraction. The turnover of this compound for the native synthase was low (Table 1).
Clearly, the work demonstrates the possibility of using the acceptance of unnatural substrates by the enzymes responsible for the biosynthesis of olfactory ligands as a criterion for the product to have sufficient similarity to the natural ligand, in terms of chemical space, for activity. However, the observed reversal of behavioural activity with (S)-14,15-dimethylgermacrene D (1g) is surprising and invites further electrophysiological studies, as well as the practical development of this chemistry for new attractants. It is known that repellents such as (S)-germacrene D act as such only in an appropriate context and can, under some circumstances, be components of attractant mixtures, although repellent singly.24,25 It may also be that structural features of the molecule conferring repellency can, in the context of other molecular features, have opposite effects. Once biologically active analogues of the target ligand are identified by feeding novel substrates to the native enzyme, rational site directed mutagenesis can be used to maximise production for practical purposes.
This work was supported by the BBSRC through grants BB/H017011/1 and BB/H01683X/1. We would like to thank Dr Robert Jenkins, Cardiff University for assistance with NMR spectroscopy and GC-MS experiments.
Notes and references
- G. W. Dawson, A. Mudd, J. A. Pickett, M. M. Pile and L. J. Wadhams, J. Chem. Ecol., 1990, 16, 1779 CrossRef CAS PubMed.
- T. Itoh, K. Kudo, K. Yokota, N. Tanaka, S. Hayase and M. Renou, Eur. J. Org. Chem., 2004, 406 CrossRef CAS.
- K. Rossiter, Chem. Rev., 1996, 96, 3201 CrossRef CAS PubMed.
- M. A. Birkett, S. Al Abassi, T. Kroeber, K. Chamberlain, A. M. Hooper, P. M. Guerin, J. Pettersson, J. A. Pickett, R. Slade and L. J. Wadhams, Phytochemistry, 2008, 69, 1710 CrossRef CAS PubMed.
- T. J. A. Bruce, M. A. Birkett, J. Blande, A. M. Hooper, J. L. Martin, B. Khambay, I. Prosser, L. E. Smart and L. J. Wadhams, Pest Manage. Sci., 2005, 61, 1115 CrossRef CAS PubMed.
- I. Prosser, I. G. Altug, A. L. Phillips, W. A. Konig, H. J. Bouwmeester and M. H. Beale, Arch. Biochem. Biophys., 2004, 432, 136 CrossRef CAS PubMed.
- O. Cascón, S. Touchet, D. J. Miller, V. Gonzalez, J. A. Faraldos and R. K. Allemann, Chem. Commun., 2012, 48, 9702 RSC.
- O. Cascón, G. Richter, R. K. Allemann and T. Wirth, ChemPlusChem, 2013, 78, 1334 CrossRef.
- J.-X. Li, X. Fang, Q. Zhao, J.-X. Ruan, C.-Q. Yang, L.-J. Wang, D. J. Miller, J. A. Faraldos, R. K. Allemann and X.-Y. Chen, Biochem. J., 2013, 451, 417 CrossRef CAS PubMed.
- D. J. Miller and R. K. Allemann, Nat. Prod. Rep., 2012, 29, 60 RSC.
- J. A. Faraldos, V. Gonzalez, M. Senske and R. K. Allemann, Org. Biomol. Chem., 2011, 9, 6920 CAS.
- S. Forcat and R. K. Allemann, Org. Biomol. Chem., 2006, 4, 2563 CAS.
- S. Forcat and R. K. Allemann, Chem. Commun., 2004, 2094 RSC.
- A. Deligeorgopoulou, S. E. Taylor, S. Forcat and R. K. Allemann, Chem. Commun., 2003, 2162 RSC.
- A. Deligeorgopoulou and R. K. Allemann, Biochemistry, 2003, 42, 7741 CrossRef CAS PubMed.
- M. J. Calvert, S. E. Taylor and R. K. Allemann, Chem. Commun., 2002, 2384 RSC.
- B. T. Greenhagen, P. E. O’Maille, J. P. Noel and J. Chappell, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9826 CrossRef CAS PubMed.
- M. Seemann, G. Z. Zhai, J. W. de Kraker, C. M. Paschall and D. W. Christianson, J. Am. Chem. Soc., 2002, 124, 7681 CrossRef CAS PubMed.
- L. S. Vedula, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 12719 CrossRef CAS PubMed.
- Y. Yoshikuni, T. E. Ferrin and J. D. Keasling, Nature, 2006, 440, 1078 CrossRef CAS PubMed.
- R. Ambrish, K. Alper and Z. Yang, Nat. Protoc., 2010, 5, 725 CrossRef PubMed.
- C. M. Starks, K. W. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- B. Webster, T. Bruce, S. Dufour, C. Birkenmeyer, M. Birkett, J. Hardie and J. A. Pickett, J. Chem. Ecol., 2008, 34, 1153 CrossRef CAS PubMed.
- B. Webster, T. Bruce, J. Pickett and J. Hardie, Anim. Behav., 2010, 79, 451 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (a) Material synthesis, characterisation; (b) protein preparation, purification, site directed mutagenesis; (c) enzyme incubations, kinetics; (d) electrophysiology and behavioural assays. See DOI: 10.1039/c5cc01814e |
| This journal is © The Royal Society of Chemistry 2015 |