
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural snapshots of the SCR reaction mechanism on Cu-SSZ-13†
Tobias
Günter
a,
Hudson W. P.
Carvalho
a,
Dmitry E.
Doronkin
ab,
Thomas
Sheppard
 a,
Pieter
Glatzel
c,
Andrew J.
Atkins
d,
Julian
Rudolph
e,
Christoph R.
Jacob
de,
Maria
Casapu
a and
Jan-Dierk
Grunwaldt
*ab
a,
Pieter
Glatzel
c,
Andrew J.
Atkins
d,
Julian
Rudolph
e,
Christoph R.
Jacob
de,
Maria
Casapu
a and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology, D-76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology, D-76344 Eggenstein-Leopoldshafen, Germany
cEuropean Synchrotron Radiation Facility, F-38043 Grenoble Cedex, France
dCenter for Functional Nanostructures and Institute of Physical Chemistry, Karlsruhe Institute of Technology, D-76131 Karlsruhe, Germany
eInstitute of Physical and Theoretical Chemistry, TU Braunschweig, D-38106 Braunschweig, Germany
First published on 29th April 2015
Abstract
The structure of copper sites in Cu-SSZ-13 during NH3-SCR was unravelled by a combination of novel operando X-ray spectroscopic techniques. Strong adsorption of NH3 on Cu, its reaction with weakly adsorbed NO from the gas phase, and slow re-oxidation of Cu(I) were proven. Thereby the SCR reaction mechanism is significantly different to that observed for Fe-ZSM-5.
Selective catalytic reduction of NOx by ammonia (NH3-SCR) over iron and copper based catalysts is presently the predominant way to remove hazardous NOx from automotive exhaust gases.1 In particular the chabazite-based catalysts Cu-SSZ-13 and Cu-SAPO-34 have recently attracted strong interest due to their outstanding performance and hydrothermal stability.2 Although Cu-SSZ-13 has already been commercialized, the reaction mechanism and the structure of the active sites are still strongly debated.3 Typically, single Cu2+ sites are located close to a six- or eight-member ring (6MR or 8MR).4 They are mobile, dynamically change their structure,4c and less active Cu dimers can be formed at intermediate temperatures.5 This structural variation inevitably requires operando studies.
In situ X-ray absorption spectroscopy (XAS) has uncovered high redox dynamics of Cu sites in the chabazite framework, particularly promoted during the standard SCR process.4b,6 This redox behavior, which is similar to Fe-zeolites, originates from the reaction of NO and NH3.7 The reduction of Cu2+ sites was observed also in an NH3 containing stream.7,8 Nevertheless, the nature of adsorbed species and the sequence of reaction steps are still controversial. For large-pore zeolites the reaction between gaseous or oxidatively adsorbed NO and adsorbed NH3 has been proposed.6a,9 Oxidative adsorption of NOx together with stronger non-dissociative adsorption of NH3 on Cu sites has been reported on both Cu-SAPO-34 and Cu-SSZ-13.10 Furthermore, formation of nitrosyl groups NO+ has been found by IR spectroscopy.11 The interaction of Cu2+ sites in Cu-SAPO-34 with both NO and NH3 was further evidenced by in situ EPR.12 With respect to the influence of other gaseous species, a positive effect of water for the SCR reaction has been reported, although it strongly inhibits NO oxidation.3,13 However, most of the studies that can evidence the interaction between the adsorbates and copper were not performed under operating conditions, which appears particularly important as previously demonstrated by Ribeiro et al.4b,14a Recent investigations have shown that combined synchrotron-based hard-X-ray photon-in/photon-out techniques15 allow probing in situ not only the oxidation state and the coordination sphere but also the nature of the ligands.8,14b
Here we report on the use of High Energy Resolution Fluorescence Detected XAS (HERFD-XAS) and Valence-to-Core X-ray Emission Spectroscopy (V2C XES) at the Cu K-edge under NH3-SCR operating conditions to shed light on the standard SCR mechanism over Cu-SSZ-13. For this purpose, we prepared a well-defined Cu-SSZ-13 catalyst and studied the structure at relevant SCR conditions and systematically under 12 defined reference conditions to understand the influence of each species and their interactions. The spectra were further interpreted using reference compounds and density-functional theory (DFT) calculations. In addition, we compare the results to our recent HERFD-XAS/XES study of Fe-ZSM-5.14b
The 1.2 wt% Cu-SSZ-13 catalyst was prepared by ion-exchange with copper acetate (ESI†) resulting in a Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al-ratio of 16:1 and Cu:Al = 0.2:1. EXAFS data analysis shows mostly isolated 4-fold coordinated Cu2+ sites with a Cu–O bond distance around 1.96 Å (ESI,† Fig. S3). This is in line with literature, where isolated sites were observed for the used ion exchange level, possibly close to 8 membered rings under the applied reaction conditions.16 This catalyst sample demonstrated typical SCR activity with a seagull shaped light-off curve (ESI,† Fig. S2). Next, the catalyst was tested in an operando fixed-bed quartz capillary microreactor cell.17 These experiments were performed with a sieve fraction of 100–200 μm (ESI†) to avoid mass transfer limitations,17 which can strongly influence SCR activity over Cu-zeolites.18 The suitability of the approach is underlined by the results in Fig. 1 as the conversion of NO in the operando cell was close to that in the laboratory fixed-bed reactor and water enhanced the performance as known for Cu-SCR catalysts.13,19
:Al-ratio of 16:1 and Cu:Al = 0.2:1. EXAFS data analysis shows mostly isolated 4-fold coordinated Cu2+ sites with a Cu–O bond distance around 1.96 Å (ESI,† Fig. S3). This is in line with literature, where isolated sites were observed for the used ion exchange level, possibly close to 8 membered rings under the applied reaction conditions.16 This catalyst sample demonstrated typical SCR activity with a seagull shaped light-off curve (ESI,† Fig. S2). Next, the catalyst was tested in an operando fixed-bed quartz capillary microreactor cell.17 These experiments were performed with a sieve fraction of 100–200 μm (ESI†) to avoid mass transfer limitations,17 which can strongly influence SCR activity over Cu-zeolites.18 The suitability of the approach is underlined by the results in Fig. 1 as the conversion of NO in the operando cell was close to that in the laboratory fixed-bed reactor and water enhanced the performance as known for Cu-SCR catalysts.13,19
 | ||
| Fig. 1 Catalytic activity of 1.2 wt% Cu-SSZ-13 measured in the lab fixed-bed reactor and the operando cell at the beamline using 1000 ppm NO, 1000 ppm NH3, 10% O2 and 0–1.5% H2O (GHSV 200000 h−1, further details in the ESI†). | ||
The results obtained with XAS/XES are first described separately and afterwards correlated to obtain a coherent picture. The numbering of the conditions was chosen to present spectral differences and is not according to the experimental order. Before each experiment, the sample was treated to reach its initial state.
HERFD-XANES spectra were collected during NO and NH3 oxidation, and under standard SCR with or without water (Fig. 2). At first glance, major changes of the XANES profiles are visible for conditions involving NH3. However, important variations could be observed for all investigated conditions. The HERFD-XANES spectrum of the untreated sample in synthetic air at room temperature (condition 1) contains features typical for Cu-SSZ-13:8 a pre-edge feature A at 8977.4 eV (1s → 3d transition in Cu2+),20 a small shoulder B at 8982.7 eV (1s → 4p transition of two-coordinated Cu+ sites)6a and a second shoulder C at 8986.5 eV (Cu2+ 1s → 4p transition with ligand to metal charge transfer (LMCT, shakedown)).8,21 A similar profile was recorded at 200 °C in the presence of water (cond. 4) which corresponds to hydrated Cu2+ sites. Dehydration by pretreatment at 550 °C (cond. 2) led to a slightly higher intensity of the pre-edge and of the features B and C. The white-line showed a sharper main feature E around 8997 eV accompanied by a shoulder D at 8993 eV, indicating a slight difference in the coordination sphere.22 Analysis of the pre-edge region indicates a majority of Cu2+ sites present under these conditions. The removal of O2 and H2O (cond. 3) led to a decrease of the pre-edge feature A suggesting autoreduction (about 60% Cu+ as estimated by its integration, Fig. 2, Table).
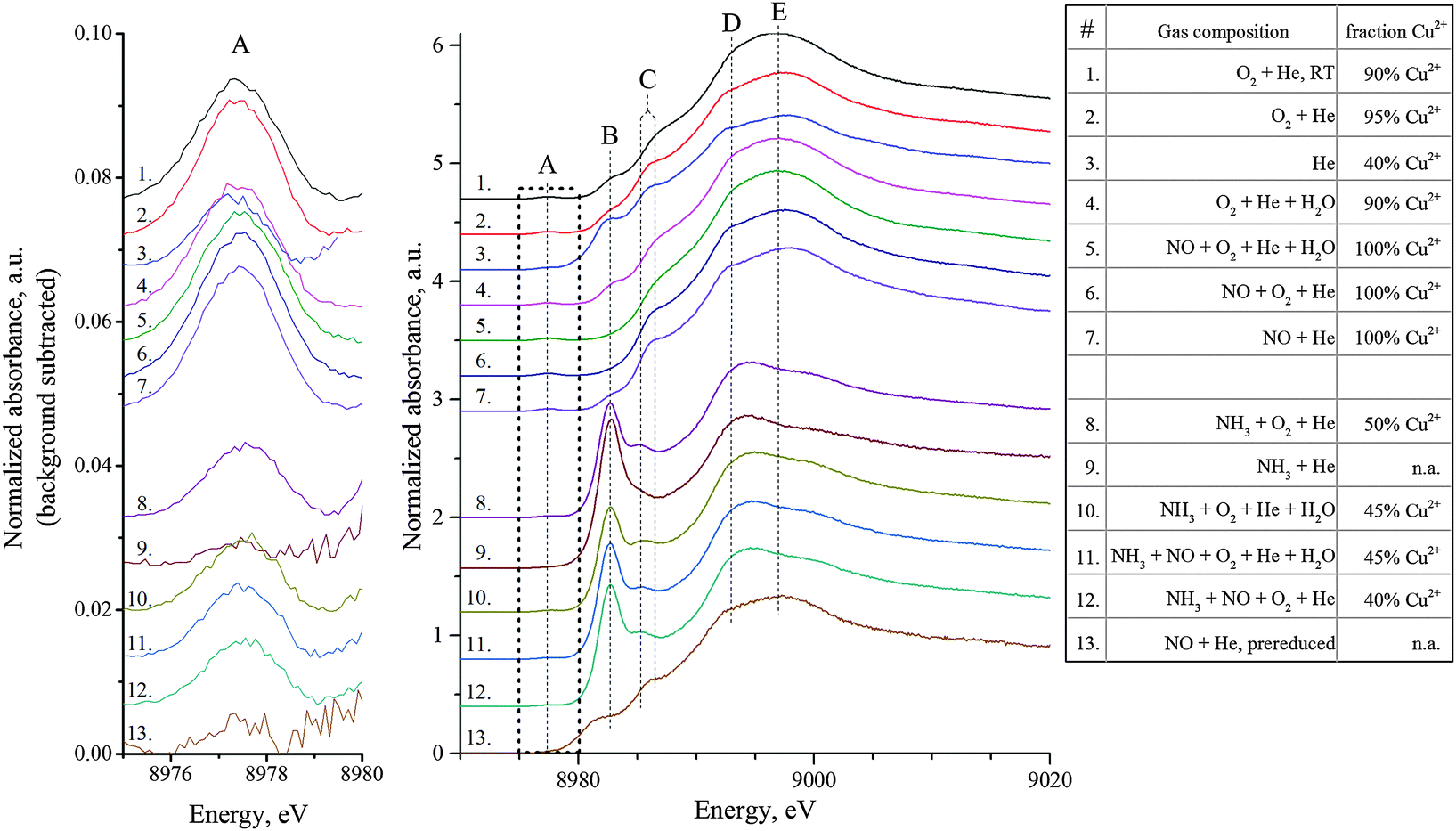 | ||
| Fig. 2 Cu K-edge HERFD-XANES (middle) and pre-edge region (left) of Cu-SSZ-13 during NH3-SCR and under model gas mixtures at 200 °C (exact gas composition and temperature given in the ESI,† Table S2). Estimation of Cu2+ amount (±10%) relative to all Cu in the sample based on the integral of the background-subtracted pre-edge peak (at 8977.4 eV) taking the spectra of conditions 1, 2, 4–7 as Cu2+ reference (details, cf. ESI†). | ||
With respect to the reactant gases, NO addition mainly affected features B and C. Thus dosing only NO + He (cond. 7) yielded the highest intensity of C but a diminishment of feature B in comparison to cond. 2 (O2/He). Both features B and C decrease when NO and O2 are dosed over the catalyst bed, indicating the complete absence of Cu+ species. This could be caused by oxidation of the remaining Cu+ sites by traces of NO2. Moreover, shoulder C almost vanishes upon addition of H2O. These results preclude the reduction of the Cu sites by NO at 200 °C. As reduction of the Cu2+ sites is expected during the SCR-reaction, the adsorption of NO on Cu+ species was verified after reduction at 550 °C in 5% H2/He (cond. 13). Note that the corresponding HERFD-XANES shows a shoulder around 8981 eV probably due to overreduction to Cu0.22,23
The second group of spectra, which involve NH3 (cond. 8–12), exhibit an intense feature B suggested to originate from linear coordination to the Cu sites.8,21,24 The highest intensity of feature B was observed in NH3-He (cond. 9) combined with an almost complete disappearance of the pre-edge feature A, which strongly suggests reduction of Cu2+ species. The addition of NH3 (cond. 8, 10) to the oxidizing atmosphere also led to reduced intensity in the 1s → 3d pre-edge region, combined with a shift of this feature by 0.2 eV to higher energies, which is possibly due to structural distortion or higher ligand field splitting.25 The presence of H2O and O2 in the gas mixture led to a stepwise decrease of feature B (higher coordination, more oxidized). As no NH3 oxidation is observed at this temperature, a reaction can be excluded, suggesting a competitive (or three-fold) adsorption of NH3 and H2O at the Cu-sites as also observed by NH3-TPD (ESI,† Fig. S16) and shown for other Cu-systems.26
Dosing the standard SCR gas mixture NO + NH3 + O2 (cond. 12, 18% NOx conversion) resulted in a spectrum similar to NH3 + O2 (cond. 8), only a little decrease of the main feature B was observed. The more realistic gas mixture with water (cond. 11) led to a spectrum similar to NH3 + O2 + H2O (cond. 10).
Next, we focused on the X-ray emission spectra (Fig. 3). Similarly to the HERFD-XANES results, the spectra can be divided into two groups showing similar features, i.e. catalyst without NH3 in the feed (cond. 1–7, 13) and catalyst with NH3 in the feed (cond. 8–12, including SCR). The differences between those groups are the following: (a) appearance of a second peak upon NH3 addition in the Kβ” region at 8958.3 eV which is seen either as broadening of peak 8956.9 eV (both peaks have similar intensity in cond. 9–11) or a shift of this peak (dominating over the peak at 8956.9 eV in cond. 8 and 12). These features are similar to those reported during in situ adsorption of NH3 on Cu-SSZ-13 by Giordanino et al.8 and can be attributed to an N atom in the coordination sphere of Cu, i.e. direct adsorption of NH3. DFT-calculations on the influence of the ligand environment on the XES spectra support such an assignment (ESI,† Fig. S10). No feature stemming from ammonia adsorbed via hydroxyl groups was detected for copper. This is in contrast to the Fe–O+H–NHx moiety observed for Fe-ZSM-5.14b (b) A peak at 8972.3 eV in the Kβ2,5 region which is shifted to 8973.4 eV and becomes more intense upon removal of reactants with only NH3/He remaining, (c) increased intensity of features at 8981.6 and 8990.0 eV after adsorption of NH3, probably due to a change in geometry of the Cu complex. The changes named above stem only from interaction with ammonia but not from the change of Cu oxidation state which is verified by exp. 13 (vs. exp. 7) where no significant changes are observed after NO-adsorption on pre-reduced Cu.
 | ||
| Fig. 3 XES spectra of Cu-SSZ-13 catalyst during NH3-SCR and under related model gas mixtures at 200 °C (exact composition and temperature given in the ESI,† Table S2). | ||
Mainly minor changes in relative intensity of the Kβ2,5 peaks were observed for the group of spectra recorded in NH3-free atmosphere. The addition of NO (in He) caused broadening or diminishment of the Kβ” features compared to the dehydrated catalyst in He. However, no clear peak at 8958.3 eV from an N atom was noted upon adsorption of NO (cond. 7). In the presence of O2 and H2O, the peak at 8956.9 eV showed basically no change upon NO addition. This indicates a strong inhibition effect particularly due to H2O (ESI,† Fig. S12, ref. 13) and points to weak adsorption of NO probably via the O atom as isonitrosyl, accompanied by small changes in the coordination environment of Cu sites.
Strikingly different from the case of NO and NH3 adsorption over Fe-ZSM-5 is the absence of a Kβ” peak at low energy which we ascribed to a positively polarized/triple coordinated oxygen atom with NO bound via Fe–O.14b This difference in adsorption of NO on Fe- and Cu-zeolites is substantial and may explain the debated differences in SCR performance and also in NO oxidation activity of those zeolites.27 Indeed, NO adsorbs via an additional O atom as a nitrite-like intermediate on an Fe site and then can be readily desorbed as NO2, whereas in the case of Cu NO seems to be adsorbed as a nitroso-group and, hence, can desorb easily again as NO or react with adsorbed NH3. DRIFTS has recently evidenced NO+ on Cu–CHA catalysts.4c,13
In summary, we observed strong NH3 adsorption on Cu sites under all conditions involving ammonia, the intensity of the NH3-related XANES features was lower for the feeds containing water. The positive influence of H2O evidenced also earlier5,19,29 can be caused either by inhibition of NH3 adsorption or by an increased Cu+ reoxidation rate.28 Concerning NO, its adsorption is supported by the change of shape of the Kβ” peak and also of the HERFD-XANES spectra (cond. 7 vs. 3). However, in the presence of O2 and/or H2O (cond. 5 and 6) only the relative intensities of peaks in the Kβ2,5 region changed and the NO adsorption must be weak. Those small changes are completely indiscernible behind the features arising from NH3 and therefore in the current state of work we assume that either NO is weakly adsorbed, or rapidly reacts from the gas phase with adsorbed ammonia/amino groups.
The proposed mechanism depicted in Scheme 1 only partially supports the mechanism suggested by Gao et al.3 since the formation of NH3–Cu+–NO+ could not be unambiguously demonstrated. Thus, the presented data indicate that during standard SCR, NH3 adsorbs on a Cu2+ site via direct coordination. Next, NO may adsorb on the same site via O and react with NH3 or directly react from the gas phase as suggested by several previous studies.6a,9,30 Both paths could circumvent the blockage of the Cu sites at low temperatures by NH3, which is in contrast to the Fe-ZSM-5 case, where the strong NH3-adsorption (ESI,† Fig. S15) additionally inhibits the reoxidation. NO might be activated by direct interaction with NH3 or weakly adsorbed via Cu-O. The decomposition of the formed nitrosoamide3 intermediate may be facilitated by the presence of water (Fig. 1) via an ammonium nitrite formation and subsequent thermal decomposition (not shown) or because it promotes the mobility and stabilizes the resulting undercoordinated Cu+. As a result of SCR and its electron transfer processes, a reduced Cu+ site with low coordination number is found, in accordance with the spatially-resolved XAS study of Cu-SAPO-34,7 which is then oxidized by O2 (no proof of NOx participation was found) to Cu2+ as a slow and rate-limiting step and stabilized by H2O as Cu2+–OH.4c In contrast, for Fe-ZSM-5 coordination of NO and NH3via Cu-O has been suggested which could not be found for Cu in Cu-SSZ-13.
 | ||
| Scheme 1 Role of copper in Cu-SSZ-13 suggested by operando XES and XAS during NH3-SCR. Possible additional water–ammonia ligands are left out for clarity. ZO stands for zeolite cation-exchange site. | ||
By combining operando HERFD-XAS and V2C XES we could provide important new insight into the structure of copper and its interaction with NH3 and NO during standard SCR. A significant difference in the intermediate species during SCR over Fe-ZSM-5 and Cu-SSZ-13 was found. This difference could explain the higher activity of Cu-zeolites at low temperatures and the absence of an NH3 inhibition effect. Furthermore, it could be correlated with the different NO oxidation behavior of the Fe- and Cu-zeolites. In future, it would be valuable to extend these studies to V-SCR catalysts and exploit the same approach of combining operando HERFD-XANES, XES and DFT calculations.
This work has been supported by KIT, the Federal Ministry of Education and Research (BMBF, projects 05K10VKB and 05K13VK2) and DBU (T. Günter). We thank ESRF for providing the beamtime at the ID26 beamline and financial support at ESRF and C. Lapras for the technical support. We are also grateful to E. Japke for the assistance during XES measurements and T. Bergfeldt (IAM-AWP, KIT) for elemental analysis.
Notes and references
- (a) I. Nova and E. Tronconi, Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, Springer, New York, 2014 Search PubMed; (b) U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413 CrossRef CAS.
- (a) P. G. Blakeman, E. M. Burkholder, H.-Y. Chen, J. E. Collier, J. M. Fedeyko, H. Jobson and R. R. Rajaram, Catal. Today, 2014, 231, 56 CrossRef CAS PubMed; (b) S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. F. Peden and D. H. Kim, Catal. Today, 2012, 184, 252 CrossRef CAS PubMed.
- F. Gao, J. Kwak, J. Szanyi and C. H. F. Peden, Top. Catal., 2013, 56, 1441 CrossRef CAS.
- (a) F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20 CrossRef CAS PubMed; (b) S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87 CrossRef CAS PubMed; (c) R. Zhang, J.-S. McEwen, M. Kollár, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, ACS Catal., 2014, 4, 4093 CrossRef CAS.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1 CrossRef CAS PubMed.
- (a) C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828 CrossRef CAS PubMed; (b) J. S. McEwen, T. Anggara, W. F. Schneider, V. F. Kispersky, J. T. Miller, W. N. Delgass and F. H. Ribeiro, Catal. Today, 2012, 184, 129 CrossRef CAS PubMed.
- D. E. Doronkin, M. Casapu, T. Günter, O. Müller, R. Frahm and J.-D. Grunwaldt, J. Phys. Chem. C, 2014, 118, 10204 CAS.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552 CrossRef CAS.
- H. Sjövall, E. Fridell, R. Blint and L. Olsson, Top. Catal., 2007, 42–43, 113 CrossRef.
- (a) L. Shi, T. Yu, X. Wang, J. Wang and M. Shen, Acta Phys.-Chim. Sin., 2013, 29, 1550 CAS; (b) H. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16 CrossRef CAS PubMed.
- J. Szanyi, J. H. Kwak, H. Zhu and C. H. F. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368 RSC.
- T. Yu, T. Hao, D. Fan, J. Wang, M. Shen and W. Li, J. Phys. Chem. C, 2014, 118, 6565 CAS.
- M. P. Ruggeri, I. Nova, E. Tronconi, J. A. Pihl, T. J. Toops and W. P. Partridge, Appl. Catal., B, 2015, 166–167, 181 CrossRef CAS PubMed.
- (a) V. F. Kispersky, A. J. Kropf, F. H. Ribeiro and J. T. Miller, Phys. Chem. Chem. Phys., 2012, 14, 2229 RSC; (b) A. Boubnov, H. W. P. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J.-D. Grunwaldt, J. Am. Chem. Soc., 2014, 136, 13006 CrossRef CAS PubMed.
- (a) P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65 CrossRef CAS PubMed; (b) N. Lee, T. Petrenko, U. Bergmann, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2010, 132, 9715 CrossRef CAS PubMed; (c) M. Bauer, Phys. Chem. Chem. Phys., 2014, 16, 13827 RSC.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548 RSC.
- J. D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037 RSC.
- P. S. Metkar, V. Balakotaiah and M. P. Harold, Chem. Eng. Sci., 2011, 66, 5192 CrossRef CAS PubMed.
- H. Sjövall, L. Olsson, E. Fridell and R. J. Blint, Appl. Catal., B, 2006, 64, 180 CrossRef PubMed.
- E. M. C. Alayon, M. Nachtegaal, E. Kleymenov and J. A. van Bokhoven, Microporous Mesoporous Mater., 2013, 166, 131 CrossRef CAS PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433 CrossRef CAS.
- N. B. Castagnola, A. J. Kropf and C. L. Marshall, Appl. Catal., A, 2005, 290, 110 CrossRef CAS PubMed.
- M. K. Neylon, C. L. Marshall and A. J. Kropf, J. Am. Chem. Soc., 2002, 124, 5457 CrossRef CAS PubMed.
- R. A. Himes, G. Y. Park, A. N. Barry, N. J. Blackburn and K. D. Karlin, J. Am. Chem. Soc., 2007, 129, 5352 CrossRef CAS PubMed.
- K.-I. Shimizu, H. Maeshima, H. Yoshida, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 862 RSC.
- (a) P. Kappen, J.-D. Grunwaldt, B. S. Hammershøi, L. Tröger and B. S. Clausen, J. Catal., 2001, 198, 56 CrossRef CAS; (b) R. Kumashiro, Y. Kuroda and M. Nagao, J. Phys. Chem. B, 1998, 103, 89 CrossRef.
- M. Ruggeri, I. Nova and E. Tronconi, Top. Catal., 2013, 56, 109 CrossRef CAS.
- G. T. Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064 CrossRef CAS.
- (a) J. A. Sullivan, J. Cunningham, M. A. Morris and K. Keneavey, Appl. Catal., B, 1995, 7, 137 CrossRef CAS; (b) A. V. Salker and W. Weisweiler, Appl. Catal., A, 2000, 203, 221 CrossRef CAS.
- L. Xu, R. W. McCabe and R. H. Hammerle, Appl. Catal., B, 2002, 39, 51 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis, catalytic performance, EXAFS, DFT calculations. See DOI: 10.1039/c5cc01758k |
| This journal is © The Royal Society of Chemistry 2015 |