
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of NAD(P)H-dependent enzyme activity with dynamic luminescence quenching of terbium complexes†
Hiroki
Ito
ab,
Takuya
Terai
*ab,
Kenjiro
Hanaoka
a,
Tasuku
Ueno
ab,
Toru
Komatsu
ac,
Tetsuo
Nagano
d and
Yasuteru
Urano
*abe
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
bCREST, JST, 7 Gobancho, Chiyoda-ku, Tokyo 102-0076, Japan
cPRESTO, JST, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dOpen Innovation Center for Drug Discovery, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
eGraduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: t-terai@mol.f.u-tokyo.ac.jp; uranokun@m.u-tokyo.ac.jp
First published on 16th April 2015
Abstract
We discovered that positively charged terbium complexes bearing 1,4,7,10-tetraazacyclododecane functionalized with amide ligands are highly sensitive to dynamic luminescence quenching by NAD(P)H. We exploited this phenomenon to establish a general time-resolved luminescence-based assay platform for sensitive detection of NAD(P)H-dependent enzyme activities.
Reduced nicotinamide adenine dinucleotide (phosphate), or NAD(P)H, is an important cofactor of oxidoreductases, which transfer electrons from NAD(P)H to their substrates, affording NAD(P)+ and the products. These enzymes function in many metabolic pathways, and are considered potential therapeutic targets.1,2 Assay of their activities is also helpful for clinical diagnosis.3 However, current assays of NAD(P)H-dependent enzyme activities are mostly based on absorption measurement, and this strategy has many drawbacks. For example, monitoring the absorption of NAD(P)H itself at 340 nm, which is a common approach, gives poor sensitivity in small volumes, such as 384- or 1536-well microplates,4 and absorbance in the UV region is influenced by many extraneous factors. Colouring of tetrazolium salts upon NAD(P)H-dependent reduction is also used in cell proliferation and toxicity assays, but chemical instability of these reagents4 limits their applicability. Fluorescence measurement of NAD(P)H at 460 nm is another option, but is rarely used in practice, partly due to poor sensitivity at low NAD(P)H concentrations and an inner filter effect at high NAD(P)H concentrations.4 Although a few chemodosimeters for NAD(P)H have been reported,5 they are not suitable for monitoring enzyme activity in real time. Hence, we thought that a novel strategy to detect NAD(P)H that overcomes these disadvantages would have many potential applications, especially for high-throughput screening (HTS) in drug discovery and for clinical diagnostic tests.
For this purpose, we focused on luminescent lanthanide complexes. Lanthanide (Tb3+ or Eu3+) complexes have long luminescence lifetimes of millisecond order,6 which enables sensitive detection by measuring only the long-lived component of luminescence (i.e. time-resolved measurement). By exploiting this capability, functional luminescent probes for various analytes including ions7 and enzymes8 have been developed,9 but no luminescent probes for NAD(P)H have yet been reported. Interestingly, Parker et al. recently showed that some lanthanide complexes are susceptible to dynamic quenching by a few biological reductants such as ascorbate and urate, probably via exciplex formation.10,11 Because they were interested in the use of luminescent complexes in cells, they regarded this quenching as undesirable, with one exception,12 and they developed complexes that were tolerant of reductants.10 However, we thought that we could make use of this kind of intermolecular quenching to develop a novel and general strategy to detect NAD(P)H-dependent enzyme activity. In principle, this should be feasible, because NAD(P)H is one of the most potent biological reductants (Table S1, ESI†), which would efficiently quench luminescence, whereas NAD(P)+, an oxidized form of NAD(P)H, would not. Herein, we show that Tb3+ complexes are indeed dynamically quenched by NAD(P)H and the magnitude of the quenching can be modulated by adjusting the overall charge of the complex (Fig. 1a). Based on this finding, we have developed a novel general luminescence intensity- and lifetime-based assay for NAD(P)H-dependent enzyme activities.
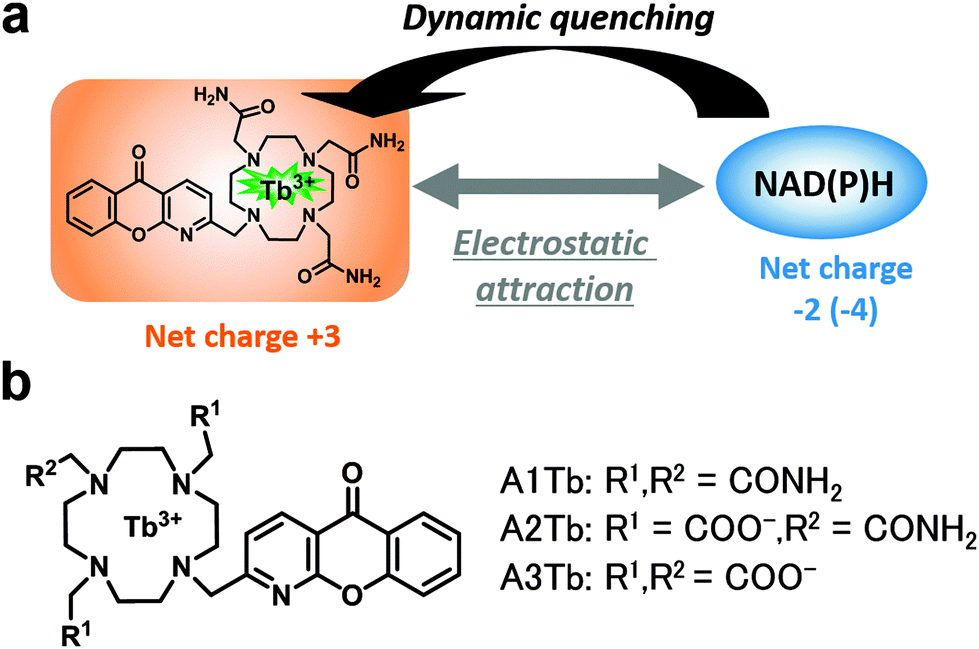 | ||
| Fig. 1 (a) Schematic representation of the interaction of the Tb3+ complex and NAD(P)H. (b) Chemical structure of complexes. | ||
We first hypothesized that the efficiency of the expected quenching by NAD(P)H would depend on the structure of the chelating moiety, in particular its charge. To test this idea, we designed and synthesized three Tb3+ complexes with different net charges (Fig. 1b). Negatively charged NAD(P)H is expected to show greater electrostatic attraction towards a more positively charged complex, as previously reported for ATP.13 As a scaffold complex, we selected A3Tb, previously reported by Parker et al.,11b,12 and prepared a series of side-chain derivatives with altered net charge, because 1,4,7,10-tetraazacyclododecane and its derivatives form kinetically and thermodynamically stable complexes with lanthanide ions.14 Three of the ring nitrogen atoms were substituted with amide and/or carboxylate groups that coordinate to Tb3+. Azaxanthone was used as an antenna moiety because it sensitizes Tb3+ with high efficiency.15 The changes in luminescence intensity and lifetime of A1Tb, the complex with the highest positive charge, upon titration of NADH are illustrated in Fig. 2. Notably, the values of both parameters were greatly decreased in the presence of as little as 1 μM NADH, and the addition of 100 μM NADH almost totally quenched the luminescence. The quenching was concentration-dependent, and the Stern–Volmer constants (Ksv) of luminescence intensity and lifetime were almost identical at low NADH concentrations (Fig. S1, ESI†), indicating that the quenching process primarily occurs in the excited state of the Tb3+.16 The data demonstrate that Tb3+ complexes with appropriate structures are indeed dynamically quenched by NADH with high efficiency. On the other hand, complexes with less positive charge and neutral charge, A2Tb and A3Tb, respectively, showed only moderate decreases in luminescence intensity and lifetime upon titration of NADH up to 100 μM (Fig. S2 and S3, ESI†).
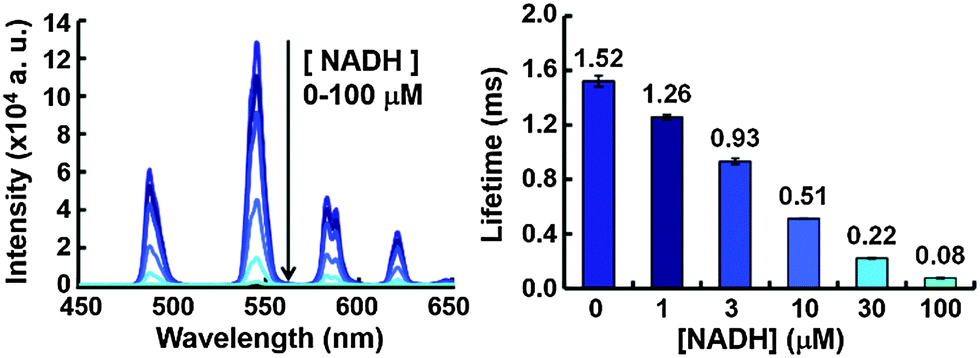 | ||
| Fig. 2 Luminescence spectra (left) and luminescence lifetime (right) of A1Tb upon titration of NADH from 0 μM (dark blue) to 100 μM (aqua). Error bars represent S.D. (n = 3). | ||
We further investigated the photophysical properties of these Tb3+ complexes and the bimolecular quenching constants (kq) upon titration of NADH. No significant differences were observed in the shapes of the absorption spectra and emission spectra, which may imply that the coordination structures of the three complexes are very similar (Fig. S4 and Table S2, ESI†). As summarized in Table 1, the more positively charged complex was more sensitive to NADH. The trend was more clearly seen in quenching of A1Tb by NADPH, which possesses greater negative charge than NADH, and is therefore expected to have stronger electrostatic interaction than NADH with positively charged compounds (compare Tables S3 and S4, ESI†). On the other hand, 100 μM NAD+ had little effect on luminescence intensity and no effect on lifetime (Table S5, ESI†). We also measured the effect of several adenine-containing biological compounds, and showed that they do not cause dynamic quenching of the complex (Table S5, ESI†). All these data suggest that electrostatic attraction between Tb3+ and phosphate groups, as well as reductive electron transfer or charge-transfer exciplex formation between Tb3+ and the reduced nicotinamide moiety at the excited state, is important for the dynamic quenching by NAD(P)H.17 However, considering the energy diagram of Tb3+ complexes (Fig. S5, ESI†), which starts with antenna excitation, followed by intersystem crossing and intramolecular energy transfer, and results in the excited state of Tb3+, it is possible that NAD(P)H also acts on the antenna moiety. Therefore, we further examined whether the excited state of the antenna is involved in the quenching process by direct excitation of the Tb3+ ion at 488 nm (Tables S3 and S4, ESI†). The obtained quenching constants were the same for direct excitation (488 nm) and antenna excitation (340 nm), supporting the idea that NAD(P)H only acts in the excited state of Tb3+. In accordance with this view, the fluorescence of the antenna moiety, 2-Me azaxanthone, was hardly quenched in the presence of 10 μM NADH (Fig. S6, ESI†). Importantly, the kq value of A1Tb for NAD(P)H (over 1 × 108 M−1 s−1) was much larger than those for GSH and ascorbate, so A1Tb is tolerant of these reductive reagents, which are often used in enzyme assay (Table S1, ESI†).
| Probe | Charge | τ (ms) | K sv (M−1) | k q (M−1 s−1) |
|---|---|---|---|---|
| a K sv and kq were calculated from Stern–Volmer plots of luminescence lifetimes upon titration of NADH. Excitation wavelength was 340 nm. | ||||
| A1Tb | +3 | 1.52 | 1.9 × 105 | 1.2 × 108 |
| A2Tb | +1 | 1.96 | 4.0 × 104 | 2.1 × 107 |
| A3Tb | 0 | 1.87 | 1.7 × 104 | 9.1 × 106 |
Next, we tested whether this quenching can be used to monitor enzyme reactions, because A1Tb responds to 1–100 μM NAD(P)H, which is the optimal range for applications such as HTS. The selected model enzyme, lactate dehydrogenase (LDH, EC 1.1.1.27), is a NADH-dependent enzyme that catalyzes the reduction of pyruvate to L-lactate while converting NADH into NAD+ (Fig. 3a); it plays a key role in the anaerobic pathway of glucose metabolism.1 Because cancer cells tend to use this pathway instead of the aerobic pathway (Warburg effect), a specific subtype of this enzyme, LDH-A, is considered as an attractive target for anticancer drugs.1 The results of LDH assay with A1Tb in a cuvette are shown in Fig. 3b. Addition of LDH was accompanied by a monotonic increase in both luminescence intensity and lifetime of A1Tb, while no change was observed in the absence of the enzyme. Unaltered LDH activity in the presence of A1Tb was confirmed by measuring NADH absorption (Fig. S7, ESI†), which indicates that the probe does not interfere with the reaction of LDH. Also, the reaction products, L-lactate and NAD+, had negligible effect on the luminescence intensity or lifetime of A1Tb, at least up to 100 μM (Table S5 and Fig. S8, ESI†). These data demonstrate that the conversion of NADH into NAD+ by LDH changed the luminescence properties of A1Tb. We next measured LDH activities on 96-well plates to see whether the method is suitable for HTS. As little as 1 mU mL−1 of LDH could be detected using A1Tb on microplates after incubation for 25 minutes. In contrast, 10 mU mL−1 was the lowest concentration that could be detected by absorption measurement of NADH at 340 nm (Fig. 3c). Thus, A1Tb provides a more sensitive measurement of enzyme activity than this classical method.
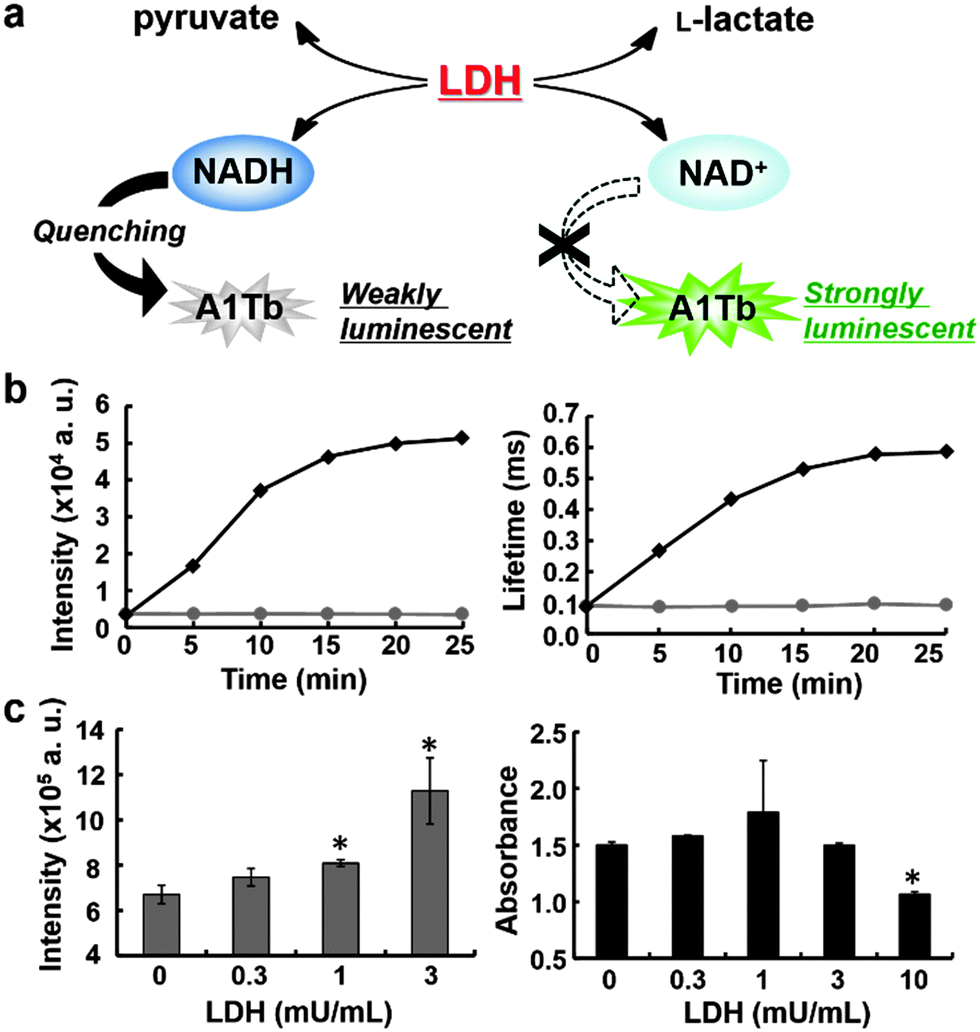 | ||
| Fig. 3 (a) Schematic illustration of LDH assay using lanthanide luminescence. (b) Time course of luminescence intensity (left) or lifetime (right) of 4 μM A1Tb in the presence (black diamond) and absence (gray circle) of LDH (50 mU mL−1). Initial concentrations of pyruvate and NADH were 125 μg mL−1 and 100 μM, respectively. (c) Comparison of time-resolved luminescence (left) and NADH absorption (right) measurements on microplates after incubation for 25 minutes. Error bars represent S.D. (n = 3). Asterisks indicate p < 0.05 vs. 0 mU mL−1. | ||
The scope of A1Tb is not limited to LDH. To demonstrate its generality, we performed preliminary time-resolved luminescence measurements for alcohol dehydrogenase (ADH, EC 1.1.1.1) and malate dehydrogenase (MDH, EC 1.1.1.37), and confirmed that A1Tb could also probe their activity on microplates with small S.D. values (Fig. S9 and S10, ESI†). In principle, our probe can monitor the activities of hundreds of NAD(P)H-dependent enzymes, and the number could be further increased by coupling these enzymes to other enzymes. A further advantage of our probe is that it is reversible (i.e. the luminescence can be turned on and off repeatedly). For example, when the NADH concentration in the sample was initially increased and then decreased, the probe could readily report these changes in real time (Fig. S11, ESI†). This is in sharp contrast to reaction-based fluorescence and luminescence sensors.5
In conclusion, we have developed a novel assay platform for NAD(P)H-dependent enzymes using luminescent Tb3+ complexes. Though many luminescent probes for biological molecules have been reported,7–9,12 most of them are specific to limited analytes and do not have practical utility for general assays. In contrast, probes for coenzymes13 including NAD(P)H are expected to have high generality. We believe that this is the first report to describe the interaction between NAD(P)H and luminescent lanthanide complexes.17 We showed that NAD(P)H dynamically quenches the luminescence of Tb3+ complexes, and this phenomenon can be used to monitor the activities of NAD(P)H-dependent enzymes. Although it is not suitable for imaging cellular NADH, our probe can monitor enzyme reactions in real time with good sensitivity. In this communication, we have focused on optimizing the net charge of the complexes, and the influence of other factors still needs to be studied to elucidate the quenching mechanism in more detail. We are currently synthesizing Tb3+ complexes with other antenna or chelator moieties to optimize sensitivity, and we aim to apply them for inhibitor screening and clinical diagnostic assays of NAD(P)H-dependent enzymes.
This work was supported by KAKENHI (Grant 22000006 to T.N., 24689003 and 24659042 to K.H., 24655147 to K.T., 25104506 to T.T., and 23249004 to Y.U.), by SENTAN, JST (K.H.), Grant-in-Aid for JSPS Fellows (H.I.) and by Astellas Foundation for Research on Metabolic Disorders (T.T.).
Notes and references
- M. G. Vander Heiden, Nat. Rev. Drug Discovery, 2011, 10, 671 CrossRef CAS PubMed.
- (a) K. Smolková and P. Ježek, Int. J. Cell Biol., 2012, 273947 Search PubMed; (b) M. K. Go, W. C. Zhang, B. Lim and W. S. Yew, Biochemistry, 2014, 53, 947 CrossRef CAS PubMed.
- S. L. Upstone, Encyclopedia of Analytical Chemistry, John Wiley & Sons Ltd, Chichester, 2000 Search PubMed.
- M. J. Vázquez, S. Ashman, F. Ramón, D. Calvo, A. Bardera, J. J. Martín, M. Rüdiger, D. Tew and J. M. Domínguez, J. Biomol. Screening, 2006, 11, 75 CrossRef PubMed.
- H. Komatsu, Y. Shindo, K. Oka, J. P. Hill and K. Ariga, Angew. Chem., Int. Ed., 2014, 53, 3993 CrossRef CAS PubMed.
- (a) J. Yuan and G. Wang, Trends Anal. Chem., 2006, 25, 490 CrossRef CAS PubMed; (b) I. Hemmilä, J. Biomol. Screening, 1999, 4, 303 CrossRef PubMed; (c) F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22 CrossRef CAS PubMed.
- (a) K. Hanaoka, K. Kikuchi, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2004, 126, 12470 CrossRef CAS PubMed; (b) D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520 RSC.
- (a) M. S. Tremblay, M. Halim and D. Sames, J. Am. Chem. Soc., 2007, 129, 7570 CrossRef CAS PubMed; (b) T. Terai, H. Ito, K. Kikuchi and T. Nagano, Chem. – Eur. J., 2012, 18, 7377 CrossRef CAS PubMed; (c) T. Terai, K. Kikuchi, S. Iwasawa, T. Kawabe, Y. Hirata, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2006, 128, 6938 CrossRef CAS PubMed.
- For review, see J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- R. A. Poole, C. P. Montgomery, E. J. New, A. Congreve, D. Parker and M. Botta, Org. Biomol. Chem., 2007, 5, 2055 CAS.
- (a) F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975 RSC; (b) G.-L. Law, D. Parker, S. L. Richardson and K.-L. Wong, Dalton Trans., 2009, 8481 RSC.
- R. A. Poole, F. Kielar, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084 RSC.
- (a) S. Nadella, P. M. Selvakumar, E. Suresh, P. S. Subramanian, M. Albrecht, M. Giese and R. Fröhlich, Chem. – Eur. J., 2012, 18, 16784 CrossRef CAS PubMed; (b) E. A. Weitz, J. Y. Chang, A. H. Rosenfield and V. C. Pierre, J. Am. Chem. Soc., 2012, 134, 16099 CrossRef CAS PubMed.
- X. Zhu and S. Z. Lever, Electrophoresis, 2002, 23, 1348 CrossRef CAS.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294 CrossRef CAS PubMed.
- Replacing Tb3+ with Eu3+ greatly reduced the extent of quenching, as has been reported for other reductants.10,11a.
- There is a report on the ground state reduction of the Dy3+ complex by NADH on the film ( T. Basova, I. Jushina, A. G. Gürek, V. Ahsen and A. K. Ray, J. R. Soc., Interface, 2008, 5, 801 CrossRef CAS PubMed ), but we do not believe this is directly relevant to our probes, because our probes showed no change in absorption spectra.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic protocols and characterization of compounds, photophysical properties of complexes, absorption and luminescence spectra of complexes upon addition of NADH, the effect of analogous compounds, an energy diagram of Tb3+ complexes, fluorescence spectra of the antenna moiety, additional data of LDH assay, detection of other enzymes, and the reversible nature of the method. See DOI: 10.1039/c5cc01613d |
| This journal is © The Royal Society of Chemistry 2015 |