
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum confinement-tunable intersystem crossing and the triplet state lifetime of cationic porphyrin–CdTe quantum dot nano-assemblies†
Ghada H.
Ahmed
,
Shawkat M.
Aly
,
Anwar
Usman
,
Mohamed S.
Eita
,
Vasily A.
Melnikov
and
Omar F.
Mohammed
*
Solar and Photovoltaics Engineering Research Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: omar.abdelsaboor@kaust.edu.sa
First published on 27th March 2015
Abstract
Here, we report a ground-state interaction between the positively charged cationic porphyrin and the negatively charged carboxylate groups of the thiol ligands on the surface of CdTe quantum dots (QDs), leading to the formation of a stable nanoassembly between the two components. Our time-resolved data clearly demonstrate that we can dramatically tune the intersystem crossing (ISC) and the triplet state lifetime of porphyrin by changing the size of the QDs in the nanoassembly.
Light absorption, exciton dissociation, and charge collection are considered to be the three key processes that control the overall power-conversion efficiencies of organic photovoltaic devices.1,2 Matching the exciton diffusion length with the respective optical absorption length is a challenge for achieving high power-conversion efficiencies in solar cells.2,3 In this respect, photo-generated singlet excitons exhibit very short lifetimes because of their dipole-allowed spin radiative decay, and consequently, they have short diffusion lengths.3 In contrast, the radiative decay of triplet excitons is dipole forbidden; therefore, their lifetimes are considerably longer.3 This has motivated interest in the triplet state and its impact on solar cells.2,3 Being in this regime, porphyrins, the essential chromophores within nature's most efficient energy conversion device,4 have received special attention due to their good thermal stability and remarkable photo-electrochemical properties.5,6 Driven by their potential applications in the fields of artificial photosynthesis and photovoltaics, the excited-state dynamics of porphyrins have attracted increased research attention.7,8 The electronic transitions of the Soret and Q-bands that appear in the visible spectral range are related to the symmetry of the macrocyclic moiety of porphyrins and have intense absorptions in the visible spectral region.9 The excitation of porphyrins in the Soret band leads to internal conversion to the lowest singlet excited state (S1). This conversion is followed by either decay to the ground state or by a singlet (S1)–triplet (T1) intersystem crossing (ISC) to populate the low-lying triplet state.10 The rate of the ISC process depends on both the energy gap between S1 and T1 and the spin–orbit coupling.11,12
The excited-state properties of porphyrins can be systematically adjusted by introducing distortion into the porphyrin macrocycle,13 which has triggered several interaction studies with the porphyrin tetrapyrrole macrocycle. More specifically, the interactions of porphyrins with nanostructures, such as gold nanoparticles,14 silver nanospheres,15 semiconductor QDs,16,17 metal oxide nanoparticles,18 carbon nanotubes19 and graphene,20 have also attracted considerable interest. These studies predominantly focused on distinguishing and understanding the photoinduced electron transfer from energy transfer in the porphyrin nanoassemblies. Time-resolved pump–probe spectroscopy provides crucial information for the complete understanding of the photoinduced excited state transitions,21,22 including electron and energy transfer in their nanoassemblies.23
In this study, we explore the excited state deactivation processes of 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluene-sulfonate) (TMPyP) upon the addition of water-soluble thioglycolic acid (TGA)-capped CdTe QDs (the structure is given in Scheme S1, ESI†). The four pyridinium moieties result in TMPyP being positively charged at neutral pH and can strongly bind with the negatively charged TGA groups capping the surface of the CdTe QDs through electrostatic interactions to bring the TMPyP and CdTe QD units in close proximity. Among the chalcogenide-based nanoscale materials, CdTe QDs were selected for this study due to their band gap energy in the UV-Vis region, water solubility and high affinity for porphyrins.16,17 In the electrostatic binding and nanoassembly formation of TMPyP with CdTe QDs, the rate of the ISC and the triplet state lifetime of TMPyP were found to be sensitive to the size of the QDs. For comparison purposes, we investigated the interactions between neutral 5,10,15,20-tetra(4-pyridyl)-porphyrin (TPyP) and CdTe QDs, revealing that the positive charge on the meso units is the key for controlling the photo-physical processes.
As shown in Fig. 1, the free-base TMPyP typically has an intense Soret band at 416 nm and weaker Q-bands at 515, 555, 586, and 640 nm. The successive addition of 1.3 nm QDs to a solution of TMPyP resulted in a decrease in the intensity of the Soret band, followed by the appearance of a new band at 460 nm with an isosbestic point at 436 nm, providing clear evidence for ground state equilibrium between the free TMPyP and TMPyP–CdTe QD nanoassembly. At the same time, the Q-bands transform into a simple broad band with two peaks at 586 and 640 nm. Because the Soret and Q-bands correspond to the electronic structure of the macrocyclic cavity of TMPyP,15,18 these observed spectral modifications are strong indications for interactions between TMPyP macrocyclic and CdTe QDs. In such a nanoassembly, a considerable distortion in the structure of TMPyP from its free-base structure is expected to occur due to out-of-plane displacements upon the complexation of the TMPyP cavity with the QD.18
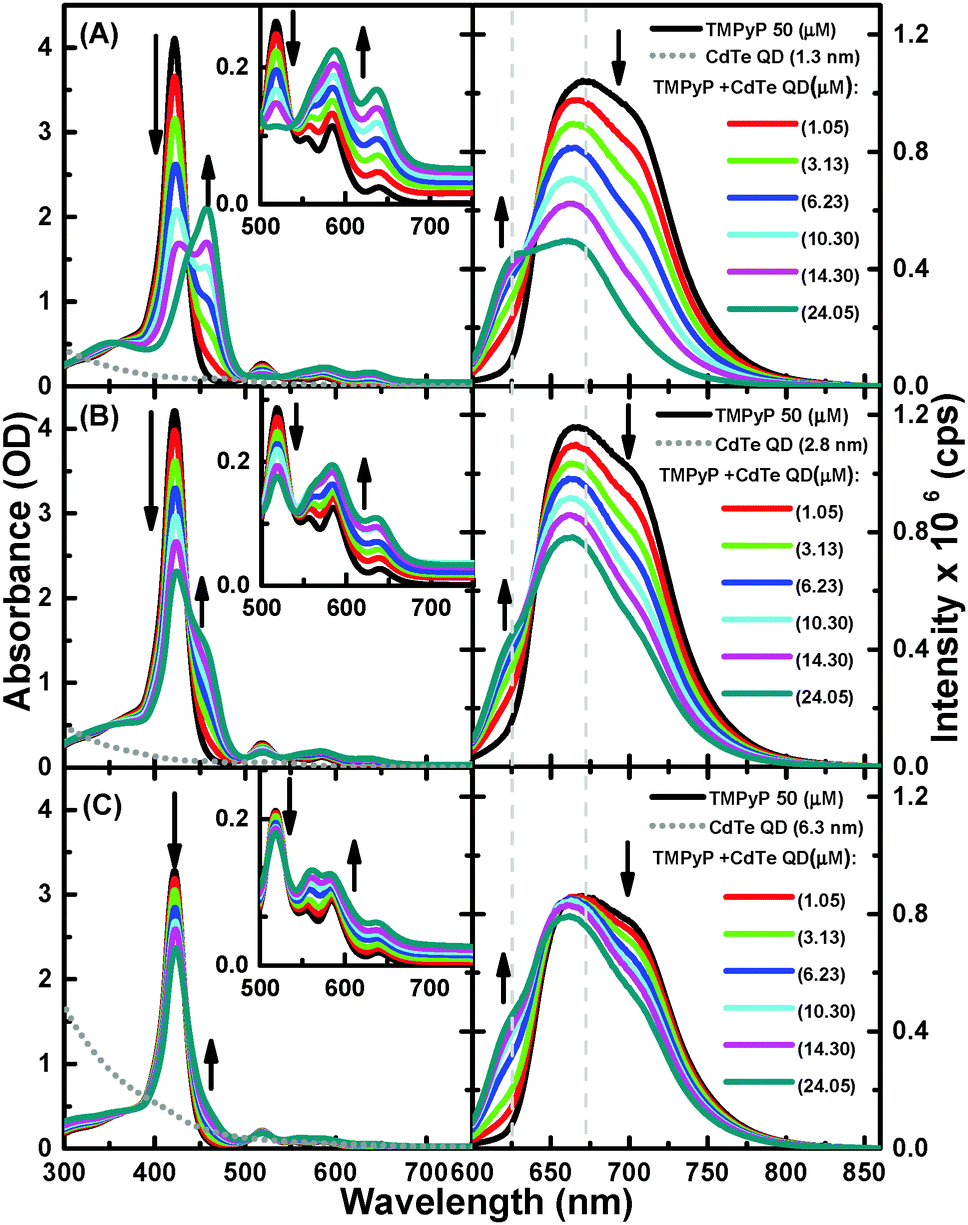 | ||
| Fig. 1 Absorption (on left) and emission (λex = 580 nm) (on right) of (A) TMPyP (50 μM) with successive additions of CdTe QDs (1.3 nm), (B) CdTe QDs (2.8 nm), and (C) CdTe QDs (6.3 nm); the concentrations are given in the figure. | ||
The formation of a TMPyP–CdTe QD nanoassembly was also indicated by quenching of the emission of TMPyP. Upon excitation at 580 nm in the absence of CdTe QDs, TMPyP exhibits a broad fluorescence band over the range of 625–800 nm, which corresponds to the S1–S0 transition. The successive addition of CdTe QDs led to a blue-shift along with a decrease in the fluorescence intensity of TMPyP (Fig. 1A). In addition, a more resolved spectrum with two peaks at 625 and 670 nm was observed, providing clear evidence for disruption of the intramolecular charge transfer (CT) between the cavity and the meso unit.24 To evaluate the dependence of the quenching behavior on the size of the QDs, we also examined the absorption and fluorescence spectra of TMPyP in the absence and presence of different sizes of CdTe QDs. We observed that the changes in both the absorption and emission spectra exhibited the same trend, but the spectral changes and emission quenching became considerably smaller as the size of the QDs increased; see Fig. 1A–C.This result is consistent with the so-called effect of the surface-to-volume ratio of the QDs, emphasizing that the charge density or quantum confinement and surface coverage of the QDs are responsible for the fluorescence quenching. In this sense, we hypothesized that the charge density should be determined by the Cd2+ ions at the surface of the QDs, which bound to the thiol ligands,25 and that surface coverage is facilitated by electrostatic interactions between the positively charged pyridinium moieties that serve as anchoring groups in TMPyP and the negatively charged carboxylic groups of the thiol ligands on the surface of the QDs.
To investigate this hypothesis, we evaluated the interaction between the CdTe QDs with neutral TPyP, a porphyrin derivative closely related to TMPyP without positively charged anchoring groups in its structure, under the same experimental conditions. As shown in Fig. S1, (ESI†) we found that the successive addition of CdTe QDs up to 14.3 μM into the TPyP solution had no effect on either the absorption or emission spectra. Thus, this finding, along with the reduction potentials of the two porphyrins (−1.01 V for TMPyP and −1.55 V for TPyP)26 and the lack of the spectral overlap between the emission of the porphyrin and the absorption of the CdTe QDs, suggests that electron and energy transfer from TPyP to CdTe has a very low probability, indicating the formation of very few nanoassemblies, if any, between the CdTe QDs and the neutral TPyP. This observation supports the hypothesis that a positive charge on the porphyrin is required to form a nanoassembly with CdTe QDs. The formation of the TMPyP–CdTe nanoassembly was also confirmed by the Raman spectra of TMPyP in the absence and presence of CdTe QDs (see SI and Fig. S2, ESI†). The number of TMPyP molecules accommodated on single QDs can be approximately estimated by calculating the surface area of the TMPyP and QDs.15 Because the interaction between TMPyP and QDs is controlled by electrostatic interactions between the peripheral pyridinium functional groups and the carboxylic group on the QD surface, we can assume that the porphyrins make a monolayer parallel to the QD surface. From the effective diameter of TMPyP of ∼2 nm15 we estimated the number of TMPyP molecules accommodated on single QDs to be 2, 8, and 40 for the QDs of 1.3, 2.8, and 6.3 nm in diameter.
Nanosecond transient absorption (ns-TA) spectra of aqueous solutions of TMPyP and TMPyP–CdTe QDs of the smallest and largest CdTe QDs (1.3 and 6.3 nm) are presented in Fig. 2. It is worth mentioning that T1–Tn absorption spectra (450–650 nm range) were collected after optical excitation at 355 nm, which is the third harmonic generation of the 1064 nm Nd:YAG Q-switched laser. More details about setups are given in the Experimental section. This observation is in excellent agreement with the literature for the transient triplet state absorption.27,28 Although there is an overall spectral similarity for the T1–Tn absorption spectra of free TMPyP in Fig. 2A to that of TMPyP–CdTe (6.3 nm) and TMPyP–CdTe (1.3 nm) nanoassemblies in Fig. 2B and C, respectively, a significant shift in the spectra to higher wavelengths as the free TMPyP transitions to its complex (nanoassembly) with the CdTe QDs can be observed. A similar shift in T1–Tn absorption was reported when going from free-base porphyrin to metallated porphyrins.29 This is also in line with the higher energy shift of the metallo-porphyrin triplet state compared to free-base ones.28 Moreover, we have conducted a control experiment monitoring the T1–Tn absorption change of TMPyP after metallation using CdCl2 and a clear spectral shift is observed (see Fig. S3, ESI†), supporting our proposed mechanism. In other words, the observed shift can be attributed to the interaction with CdTe QDs rather than spectral overlap with ground state bleach. Moreover, the magnitude of this shift, in Fig. 2, is found to be dependent on the size of QDs used for the interaction with TMPyP. Although a shift of 6 nm is observed in the TMPyP mixture with 6.3 nm CdTe QDs, a larger shift of approximately 20 nm is observed for 1.3 nm CdTe QDs.
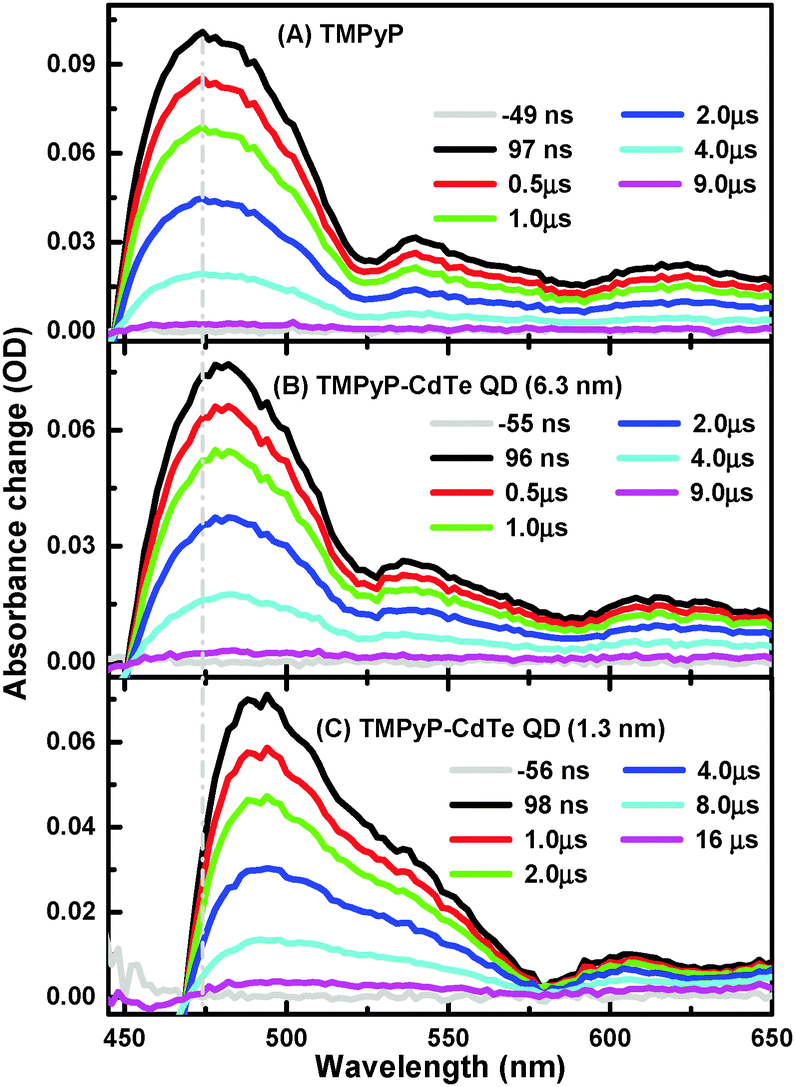 | ||
| Fig. 2 Nanosecond TA spectra of TMPyP (A) and of TMPyP in the presence of CdTe QDs (6.3 nm) (B) and CdTe QDs (1.3 nm) (C) collected after excitation of 355 nm. An identical absorption spectrum of porphyrin solutions before and after TA measurements excludes the possibility of any photo-degradation. | ||
This different magnitude of the shift suggests not only different positions for the T1 energy of TMPyP adopted in the complex relative to the free TMPyP but also different strengths of interaction between the TMPyP cavity and the surface of the CdTe QDs. Another interesting observation is the significant differences in the triple state lifetimes extracted from the TA decay profiles, as shown in Fig. 3.
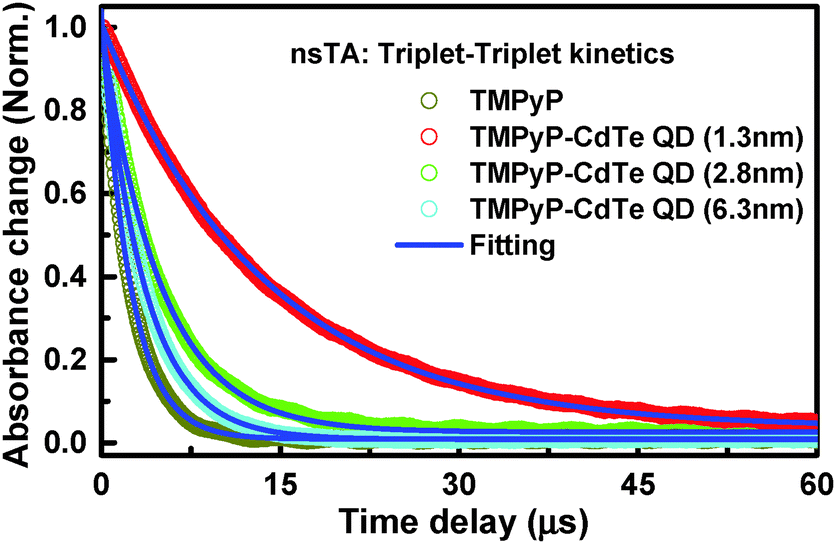 | ||
| Fig. 3 Kinetic traces of nanosecond transient absorption of the free TMPyP (grey) and TMPyP in the presence of CdTe QDs (1.3 nm; red), CdTe QDs (2.8 nm; green), and CdTe QDs (6.3 nm, cyan) collected after excitation at 355 nm. | ||
Free TMPyP exhibited a lifetime of 2.16 μs, which is approximately 6 times shorter than those extracted from the signal collected in the presence of the smallest CdTe QDs. Moreover, the lifetime becomes longer as the size of the QDs is decreased: 3.23, 5.43, and 12.25 μs in the nanoassemblies of TMPyP with CdTe QDs with sizes of 6.3, 2.8, and 1.3 nm; respectively. This observed difference is referring to the significance impact of QD's surface-to-volume ratio on the interaction with TMPyP. We anticipate, based on spectral and lifetime results, different degrees of S1–T1 mixing dependent on the change in energy levels associated with the different surface-to-volume ratios of the investigated QDs. Being in this regime, the observed longer triplet lifetime for the TMPyP–QD nanoassembly compared to TMPyP alone can be due to faster ISC from the singlet state in the nanoassemblies enhanced by virtue of spin–orbit coupling in the presence of CdTe QDs.30,31 It should be noted that upon interaction with CdTe QDs the free rotation of the meso-N-methyl pyridinium units attached to TMPyP is suppressed, which is expected to result in elongation of the excited state lifetime. In this regard, several reported examples indicate that hindered or restricted molecular rotation rigidifies the molecule and in turn reduces the non-radiative excited-state deactivation processes.32,33 It has been reported that electronic density of the triplet state is mostly located on the macrocyclic core of the porphyrin.34,35 The radiationless deactivation of the triplet state in porphyrin requires flipping of the pyridinium groups in TMPyP around the macrocyclic core of the porphyrin.34,35 Consequently, any restriction or hindrance imposed on this rotation will reduce the non-radiative deactivation of the excited state and subsequently increase the triplet state lifetime. The changes detected in the steady-state measurements indicate the adoption of the sitting-atop porphyrin complex structure.18,36 Such structural changes most likely introduced some extra restrictions on the rotation of the meso group together with the increased ISC by adopting the metallated porphyrin symmetry, as indicated by ground state measurements, leading to the observed increase in lifetime. As observed in the kinetic profiles (Fig. 3), the enhancement is strongly dependent on the size of the QDs used. Because the steady-state measurements suggested a correlation between the degrees of complexation with the size of QDs, we anticipate different degrees of ISC in the same order due to the different degrees of spin–orbit coupling by CdTe QDs, similar to the heavy atom effect.27,37,38
Further confirmation of the proposed mechanism can be obtained from the S1 lifetime determined using the TCSPC technique, and the kinetic traces are shown in Fig. S4 (ESI†). The lifetime profile extracted from the fluorescence decay of TMPyP is found to be approximately 4.01 ns, which is in good agreement with the fluorescence lifetime of TMPyP reported in the literature.29,39 On the other hand, the lifetimes obtained for TMPyP in its assembly with CdTe QDs (1.3 nm) is 0.97 ns, with CdTe QDs (2.8 nm) is 1.15 ns and with CdTe QDs (6.3 nm) is 2.45 ns, as shown in Fig. S5 (ESI†). The observed decrease in the fluorescence lifetime of TMPyP in its assemblies with QDs is pointing out to the enhancement of non-radiative routes for excited state deactivation. Since the energy and electron transfer is ruled out based on the lack of spectral overlap between TMPyP emission (which is selectively excited under our experimental conditions) and QD's absorption, and on a control experiment carried out using femtosecond TA studies using TMPyP and TMPyP–QD where no spectral signature for radical ions was detected (see Fig. S6, ESI†).
Due to the strong spectral overlap between the excited singlet and triplet state absorption, accurate values for the rate of intersystem crossing (KISC) cannot be extracted. However, we were able to calculate the KISC for TMPyP free and in the presence of CdTe QDs using the method described by Pettersson et al.,40 by which we found that the KISC is 5.9 ×107 for free TMPyP, 7.3 × 107 for TMPyP with 6.3 QD, and 16.8 × 107 for TMPyP with 1.3 QD. The increase in KISC or the decrease in lifetime can be understood in terms of the SAT or out of plane complex structure. This type of deformation can dramatically change the photophysical properties of the porphyrin.41,42 These short fluorescence lifetimes obtained for the TMPyP–CdTe QD assemblies suggest an enhancement in the ISC.
In summary, the time-resolved results demonstrate, for the first time, the possibility of modulating the ISC rate and the triplet state lifetime of TMPyP by controlling both the distance and the strength of binding between the porphyrin cavity and the surface of CdTe QDs. Fluorescence quenching of TMPyP upon addition of CdTe QDs is observed and found to be sensitive to the size of the QDs, demonstrating the impact of quantum confinement on the observed quenching. Notably, the novel insights reported in this manuscript provide an understanding of the key variables involved in the nano-assembly, thus paving the way for the exploitation of efficient ISC and subsequently elongating the triplet-state lifetime, which are required for light harvesting in the triplet state as one of potential solutions to overcome the poor exciton mobility to achieve high power-conversion efficiency.2
Notes and references
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158–162 CrossRef CAS PubMed.
- Y. Shao and Y. Yang, Adv. Mater., 2005, 17, 2841–2844 CrossRef CAS.
- C.-M. Yang, C.-H. Wu, H.-H. Liao, K.-Y. Lai, H.-P. Cheng, S.-F. Horng, H.-F. Meng and J.-T. Shy, Appl. Phys. Lett., 2007, 90, 133509 CrossRef PubMed.
- P. D. Harvey, Can. J. Chem., 2014, 92, 355–368 CrossRef CAS.
- M. Regehly, T. Wang, U. Siggel, J. H. Fuhrhop and B. Röder, J. Phys. Chem. B, 2009, 113, 2526–2534 CrossRef CAS PubMed.
- M. A. Fox, J. V. Grant, D. Melamed, T. Torimoto, C.-y. Liu and A. J. Bard, Chem. Mater., 1998, 10, 1771–1776 CrossRef CAS.
- N. M. Barbosa Neto, D. S. Correa, L. De Boni, G. G. Parra, L. Misoguti, C. R. Mendonça, I. E. Borissevitch, S. C. Zílio and P. J. Gonçalves, Chem. Phys. Lett., 2013, 587, 118–123 CrossRef CAS PubMed.
- T. Ripolles-Sanchis, B.-C. Guo, H.-P. Wu, T.-Y. Pan, H.-W. Lee, S. R. Raga, F. Fabregat-Santiago, J. Bisquert, C.-Y. Yeh and E. W.-G. Diau, Chem. Commun., 2012, 48, 4368–4370 RSC.
- G. de la Torre, P. Vázquez, F. Agulló-López and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef CAS PubMed.
- S. Perun, J. Tatchen and C. M. Marian, ChemPhysChem, 2008, 9, 282–292 CrossRef CAS PubMed.
- M. Kleinschmidt and C. M. Marian, Chem. Phys., 2005, 311, 71–79 CrossRef CAS PubMed.
- M. Kleinschmidt, J. Tatchen and C. M. Marian, J. Chem. Phys., 2006, 124, 124101 CrossRef PubMed.
- F.-Q. Bai, N. Nakatani, A. Nakayama and J.-y. Hasegawa, J. Phys. Chem. A, 2014, 118, 4184–4194 CrossRef CAS PubMed.
- A. Kotiaho, R. Lahtinen, H. Lehtivuori, N. V. Tkachenko and H. Lemmetyinen, J. Phys. Chem. C, 2008, 112, 10316–10322 CAS.
- S. Murphy, L. Huang and P. V. Kamat, J. Phys. Chem. C, 2011, 115, 22761–22769 CAS.
- M. A. Jhonsi and R. Renganathan, J. Colloid Interface Sci., 2010, 344, 596–602 CrossRef CAS PubMed.
- X. Zhang, Z. Liu, L. Ma, M. Hossu and W. Chen, Nanotechnology, 2011, 22, 195501 CrossRef PubMed.
- S. Jagadeeswari, G. Paramaguru and R. Renganathan, J. Photochem. Photobiol., A, 2014, 276, 104–112 CrossRef PubMed.
- D. M. Guldi, G. N. A. Rahman, J. Ramey, M. Marcaccio, D. Paolucci, F. Paolucci, S. Qin, W. T. Ford, D. Balbinot, N. Jux, N. Tagmatarchis and M. Prato, Chem. Commun., 2004, 2034–2035 RSC.
- S. M. Aly, M. R. Parida, E. Alarousu and O. F. Mohammed, Chem. Commun., 2014, 50, 10452–10455 RSC.
- B. Lang, S. Mosquera-Vázquez, D. Lovy, P. Sherin, V. Markovic and E. Vauthey, Rev. Sci. Instrum., 2013, 84, 073107 CrossRef PubMed.
- A. Marchioro, J. Teuscher, D. Friedrich, M. Kunst, R. van de Krol, T. Moehl, M. Gratzel and J.-E. Moser, Nat. Photonics, 2014, 8, 250–255 CrossRef CAS.
- G. P. Wiederrecht, J. E. Hall and A. Bouhelier, Phys. Rev. Lett., 2007, 98, 083001 CrossRef.
- F. J. Vergeldt, R. B. M. Koehorst, A. van Hoek and T. J. Schaafsma, J. Phys. Chem., 1995, 99, 4397–4405 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- P. Worthington, P. Hambright, R. F. X. Williams, J. Reid, C. Burnham, A. Shamim, J. Turay, D. M. Bell, R. Kirkland, R. G. Little, N. Datta-Gupta and U. Eisner, J. Inorg. Biochem., 1980, 12, 281–291 CrossRef CAS.
- A. Harriman, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1281–1291 RSC.
- E. l. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. Seixas de Melo, L. G. Arnaut, S. J. Formosinho and A. M. D. A. Rocha Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163–5169 CrossRef CAS.
- A. Harriman, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 1978–1985 RSC.
- S. P. McGlynn, T. Azumi and M. Kinoshita, Molecular Spectroscopy of the Triplet State (Prentice-Hall International Series in Chemistry), Prentice-Hall, Englewood Cliffs, New Jersey, 1969, pp. 275–276 Search PubMed.
- C. G. Claessens, A. Gouloumis, M. Barthel, Y. Chen, G. Martin, F. Agulló-López, I. Ledoux-Rak, J. Zyss, M. Hanack and T. Torres, J. Porphyrins Phthalocyanines, 2003, 7, 291 CrossRef CAS.
- S.-L. Deng, T.-L. Chen, W.-L. Chien and J.-L. Hong, J. Mater. Chem. C, 2014, 2, 651–659 RSC.
- U. Tripathy, D. Kowalska, X. Liu, S. Velate and R. P. Steer, J. Phys. Chem. A, 2008, 112, 5824–5833 CrossRef CAS PubMed.
- M. de Miguel, M. Álvaro and H. García, Langmuir, 2012, 28, 2849–2857 CrossRef CAS PubMed.
- Z. Valicsek, O. Horváth and K. Patonay, J. Photochem. Photobiol., A, 2011, 226, 23–35 CrossRef CAS PubMed.
- Y. Liang, M. Bradler, M. Klinger, O. Schalk, M. C. Balaban, T. S. Balaban, E. Riedle and A.-N. Unterreiner, ChemPlusChem, 2013, 78, 1244–1251 CrossRef CAS.
- M. Zander and G. Kirsch, Z. Naturforsch., A: Phys. Sci., 1989, 44, 205–209 CAS.
- A. S. Stasheuski, V. A. Galievsky, V. N. Knyukshto, R. K. Ghazaryan, A. G. Gyulkhandanyan, G. V. Gyulkhandanyan and B. M. Dzhagarov, J. Appl. Spectrosc., 2014, 80, 813–823 CrossRef CAS.
- K. Pettersson, K. Kilså, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2004, 126, 6710–6719 CrossRef CAS PubMed.
- K. M. Barkigia, D. J. Nurco, M. W. Renner, D. Melamed, K. M. Smith and J. Fajer, J. Phys. Chem. B, 1998, 102, 322–326 CrossRef.
- J. L. Retsek, S. Gentemann, C. J. Medforth, K. M. Smith, V. S. Chirvony, J. Fajer and D. Holten, J. Phys. Chem. B, 2000, 104, 6690–6693 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, neutral porphyrin measurement of extra TA and time-correlated single photon counting (TCSPC). See DOI: 10.1039/c5cc01542a |
| This journal is © The Royal Society of Chemistry 2015 |