
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A naphthalene diimide dyad for fluorescence switch-on detection of G-quadruplexes†
F.
Doria
a,
A.
Oppi
a,
F.
Manoli
b,
S.
Botti
a,
N.
Kandoth
a,
V.
Grande
a,
I.
Manet
*b and
M.
Freccero
*a
aDipartimento di Chimica, Università di Pavia, V. le Taramelli 10, 27100 Pavia, Italy. E-mail: mauro.freccero@unipv.it
bIstituto per la sintesi organica e la fotoreattività (ISOF), CNR, via Gobetti 101, 40129 Bologna, Italy. E-mail: ilse.manet@isof.cnr.it
First published on 24th April 2015
Abstract
A non-fluorescent naphthalene diimide (NDI) dimer, conjugating red and blue NDI dyes, becomes red/NIR emitting upon G-quadruplex binding. The fluorescence lifetime which is significantly different for the complexes, the G-quadruplex/dimer and the weakly emitting ds-DNA/dimer is the key feature for the development of new rationally engineered G-quadruplex sensors.
Naphthalene diimides (NDIs) are a very versatile platform for the design of new molecular systems able to perform a variety of functions.1 Among various potential applications of NDIs, we have focused on core-substituted NDIs as selective nucleic acid (NA) ligands and fluorescent probes. Indeed, Neidle's group and our research unit have shown that tri- and tetra-substituted NDIs are potent and reversible ligands2–4 as well as alkylating agents targeting guanine rich NAs folded into G-quadruplex (G4) structures.5–8 G-rich sequences able to fold into G4 are present in oncogene promoters9–11 as well as human telomeres and participate in biological processes crucial for cell replication and survival.10,12 Consequently, they represent a very appealing target in the development of new therapeutic approaches based on their selective recognition by multimodal molecular tools. In this context, NDIs are particularly promising. In fact, apart from their G4 affinity, their optoelectronic properties can be effectively tuned by substituents on the aromatic core,13–16 thus giving rise to absorption and emission in the red spectroscopic window which makes them appealing for fluorescence imaging and photodynamic therapy (PDT).17 In addition, the binding properties of NDIs toward G4s3,6 may also be exploited for selective photocleavage as suggested for cationic Zn-phthalocyanines.18 Although fluorescence changes upon G4 binding has been extensively investigated using small molecule ligands,19 including guanidinium-modified phthalocyanines,20 effective G4 sensing by NDIs has seldom been attempted.21 A new strategy to engineer NDIs for G4 sensing was inspired by a series of monomeric NDIs with amine substituents on the naphthalene core having excellent water solubility, good fluorescence quantum yields as well as satisfactory quantum yields for singlet oxygen production upon excitation at 640 nm.17 Here we report the synthesis and preliminary data of a water-soluble non-emitting dimeric NDI (1, Scheme 1) exhibiting a fluorescence turn-on response upon binding with specific DNA structures.
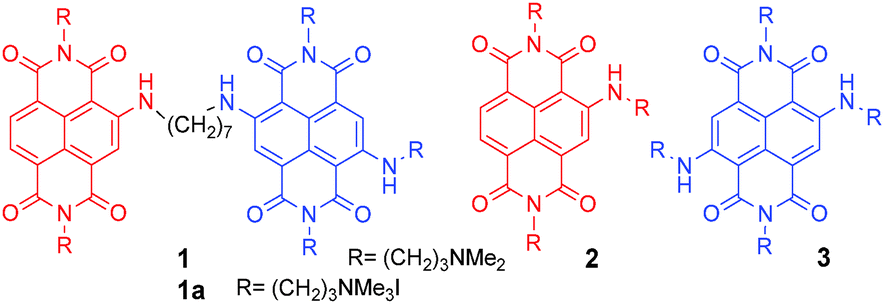 | ||
| Scheme 1 Structure of dimeric NDI 1 (resulting from the merging of monomeric NDIs 2 and 3), and its quaternary ammonium salt 1a, as iodide. | ||
1 results from the conjugation of red tri-substituted dye 2 to blue tetra-substituted NDI 3, with a (CH2)7 flexible spacer. Interestingly, time-resolved fluorescence measurements allowed differentiating between the G4 DNA complexes and ds DNA complexes of ligand 1. Dimer 1 was synthesised according to the protocol highlighted in Scheme 2. Exhaustive methylation of 1 gave the quaternary ammonium salt 1a.
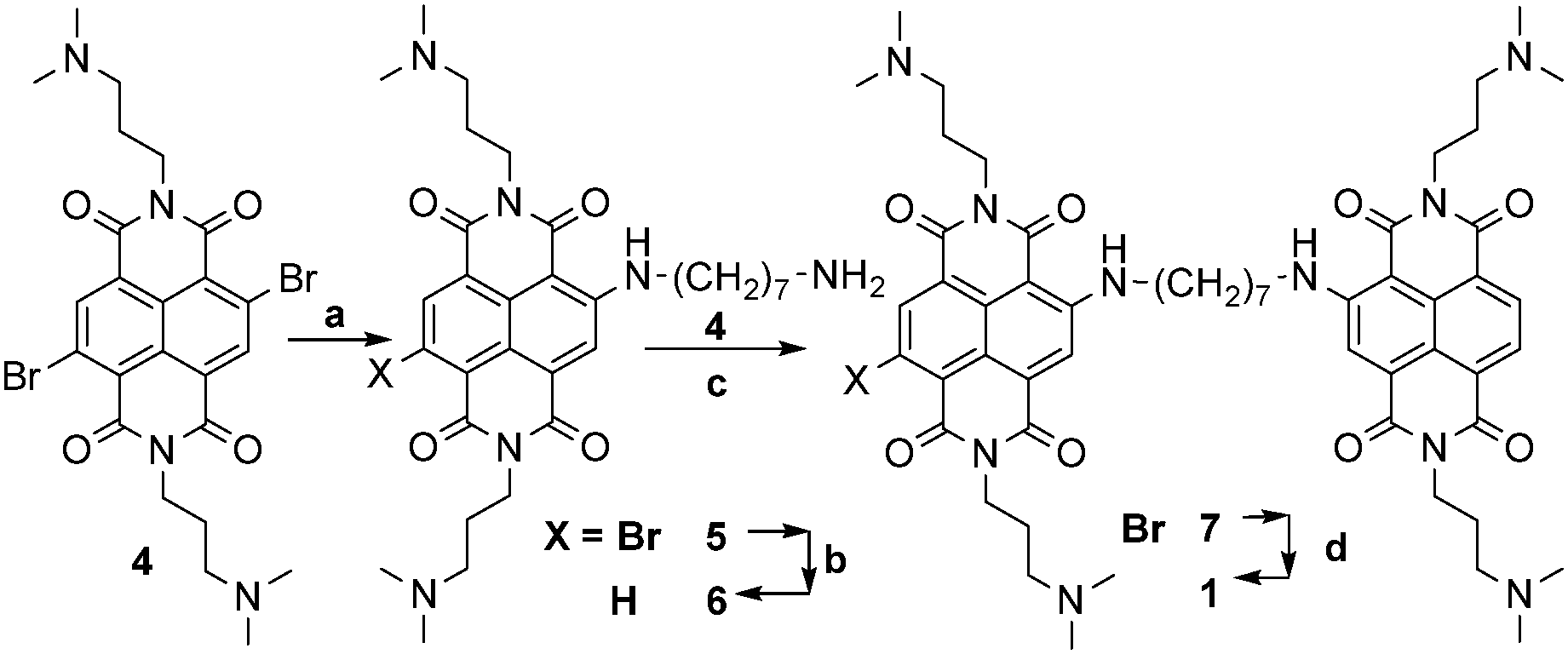 | ||
Scheme 2 Synthesis of the dimeric water soluble NDI 1. (a) 1,7-diaminoheptane 2.5 eq., CH3CN, 75 °C, 4.5 h; (b) Na2S2O4 2 eq. in aqueous CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), r.t. 1 h; (c) 0.95 eq. of 4, DMF, 50 °C, 5 h; and (d) neat N1,N1-dimethylpropane-1,3-diamine, the microwave assisted protocol, and sealed reaction vessels (M.W.; 150 °C, 200 psi, 250 bar, 200 W, 3 min). :1), r.t. 1 h; (c) 0.95 eq. of 4, DMF, 50 °C, 5 h; and (d) neat N1,N1-dimethylpropane-1,3-diamine, the microwave assisted protocol, and sealed reaction vessels (M.W.; 150 °C, 200 psi, 250 bar, 200 W, 3 min). | ||
Imidation reaction of the commercially available 2,6-dibromo-1,4,5,8-naphthalenetetracarboxylic dianhydride yielded quantitatively the 2,6-dibromo-substituted NDI 4, under acidic conditions. The subsequent nucleophilic aromatic substitution (SNAr) in the presence of an excess (2.5 eq.) of 1,7-diaminoheptane (CH3CN as solvent, 75 °C, 4.5 h) afforded a 60:40 mixture of NDIs 5 and 6 in a quantitative conversion. The lack of 5vs.6 selectivity was promptly solved by a reductive debromination step induced by Na2S2O4 in aqueous acetonitrile (1:1), which converted 5 into 6. The resulting crude was readily used for a second SNAr step on 4, using a sub-stoichiometric amount of 6. The third microwave assisted SNAr was carried out by dissolving the resulting 7 in neat N1,N1-dimethylpropane-1,3-diamine (150 °C, 200 psi, 250 bar, 200 W, 3 min, sealed reaction vessels) to give dimer 1, which crystallised from the reaction mixture. The latter protocol systematically gave rise to almost quantitative yields. Filtration, further HPLC preparative purification (CH3CN:H2O and 0.1% CF3COOH as eluent), and final anion exchange, yielded 1 as pentahydrochloride (1 × 5HCl). The protonation mode of the solubilizing amino moieties controlling both the quenching of the excited states by electron transfer (eT) and NA binding was studied potentiometrically (Fig. 1a). The remarkable acidity of fully protonated 1 (1H5, pKa1 = 2.9) and the almost overlapping pKa2 and pKa3 (7.8 and 7.9) suggest that 1 is mainly (90%) tetra-cationic (1H4) at pH 7. The monocationic (1H1) and neutral forms are populated only under basic conditions pH > 8 (pKa4 8.75, pKa5 9.12).
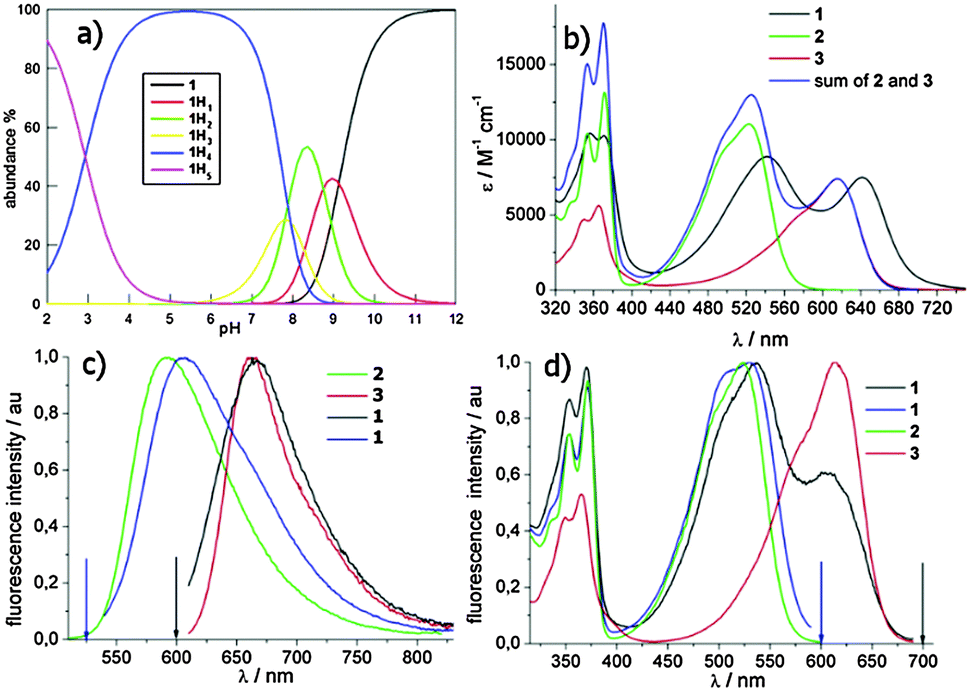 | ||
| Fig. 1 (a) Speciation analysis describing the neutral, mono-, bi-, tri-, tetra- and penta-cationic distribution of 1 (1, 1H1, 1H2, 1H3, 1H4 and 1H5) resulting from the potentiometric titrations. (b) UV-vis absorption spectra of 1, 2, 3 and the absorption sum spectrum of 2 + 3; (c) normalized corrected fluorescence spectra of 1 and its monomeric analogues 2 and 3 obtained for different excitation wavelengths indicated by the arrows; (d) normalized corrected excitation spectra of 1, 2 and 3, measured at 600 and/or 700 nm. Phosphate buffer (PB) of pH 2. | ||
1 tends to aggregate at pH > 7.8 as inferred from the UV-vis absorption titrations (ESI,† Fig. S1). Nevertheless, the absorption spectra are almost superimposable at pH ≤ 7, so the protonation state of the NMe2 groups does not significantly affect the absorption spectra. The graphs in Fig. 1 show the absorption (Fig. 1b), and corrected fluorescence spectra (Fig. 1c) as well as the fluorescence excitation spectra (Fig. 1d) of 1 in phosphate buffer at pH 2. Under these conditions, all of the aliphatic amines are fully protonated. The absorption band with vibronic signature in the 300–400 nm range is typical of the NDI core.22 The introduction of one or two amines is able to generate a second absorption band arising from a charge transfer (CT) transition involving the doublet of the aromatic amines.14,23 The absorption spectrum of 1 is clearly different from the sum of the spectra of the monomers (Fig. 1b) and displays red shifts for both absorption maxima (λmax 542/642 nm). This bathochromic shift is quite remarkable (26 nm) for the longer wavelength maximum (λmax 642 nm), which is exclusively due to the absorption of the tetra-substituted chromophore. The long and flexible spacer in the dimer likely allows strong interaction of the two aromatic cores in the ground state. Indeed, also the vibronic structure of the UV band changes markedly in dyad 1. In the presence of SDS (sodium dodecyl sulphate) micelles, the two maxima of the visible band are similar to the monomer values indicating that ground state interaction has been disrupted (ESI,† Fig. S2). 1a has a superimposable absorption spectrum.
To rationalize the photophysical behavior of the most populated form of the dimer under physiological conditions (1H4) we investigated some photophysical properties of the dimer and its monomeric homologues (2 and 3) in phosphate buffer of pH 7 and 2, where we observed the fully protonated one (1H5).
Compared to NDI 3 the fluorescence quantum yield of 1, upon exclusive excitation at 600 nm of the tetra-substituted chromophore, is very low (Table 1). A pH increase from 2 to 7 causes a small reduction of the fluorescence quantum yield (ΦF, from 0.002 to 0.001, ESI,† Fig. S3). ΦF does not change significantly passing to the quaternary ammonium salts 1a (ΦF = 0.003), suggesting a negligible effect of intramolecular electron transfer (eT) involving amine groups in the fluorescence quenching of both dyads 1 and 1a. The fluorescence lifetime (τf) measured at 690 nm for 1, similar to the fluorescence lifetime of 4.2 ns obtained for 1a, does not change with pH, so probably we are observing static quenching in the dyad. Most likely, the interaction of the two chromophores, suggested above, accounts for additional non-radiative decay pathways of the excited states in the dyad. Evaluation of the fluorescence quantum yields of the tri-substituted unit (emission peaking at 570 nm) is not straightforward due to the overlapping absorption of the tetra unit inhibiting selective excitation of the former. The fluorescence intensity of the dyad for excitation at 525 nm in buffer of pH 2 and pH 7 is nearly identical (ESI,† Fig. S4). Further, upon changing pH the fluorescence lifetimes do not change for the tri unit. Taken all together these data suggest that the protonation state of the tri-substituted chromophore does not change from pH 2 to 7, while that of the tetra-unit does. Therefore, the 1H4 species has positive charges equally distributed on both units. The excitation spectra measured at 700 nm (Fig. 1d) give some additional information on the two interacting chromophores within the dyad. Even though we cannot exclude that the tri-substituted chromophore marginally contributes to the emission at 700 nm via direct emission, the profile of the excitation spectra gives strong evidence of energy transfer from the tri-substituted unit to the tetra one, which is feasible from the energetic point of view. This is also confirmed by the excitation spectra of the dimer in the presence of SDS (ESI,† Fig. S5).24 The two lifetimes measured at 570 nm may be due to the presence of dimers in different conformations one with a short lifetime (τf = 3.35 ± 0.05 ns) and the other with a long lifetime (τf = 7.85 ± 0.05 ns), with only the former favouring energy transfer.
| NDIs | λ max (nm) | ε max (M−1 cm−1) | Φ F | τ f (ns) 570 nm | τ f (ns) 690 nm |
|---|---|---|---|---|---|
| a Fluorescence quantum yields, see ref. 17 for 2 and 3. b Fluorescence lifetime at 525 nm for excitation at 373 nm. c Fluorescence lifetime at 690 nm for excitation at 637 nm. d Fluorescence quantum yields of 0.15 and 0.13 have been reported in ref. 17 for compounds 2 and 3, respectively, at pH 7. e Exciting at 600 nm and using monomer 3 as reference. | |||||
| 2@pH 2 | 522 | 11000 |
0.19d | 5.60 | — |
| 3@pH 2 | 616 | 7400 | 0.17d | — | 4.40 |
| 1@pH 2 | 542/642 | 8870/7500 | 0.002e | 3.30, 40% | 3.93 |
| 7.80, 60% | |||||
| 1@pH 7 | 542/642 | 8870/7500 | 0.001e | 3.40, 39% | 4.02 |
| 7.90, 61% | |||||
The complexation behaviour of 1 towards four types of DNA has been studied using different spectroscopic techniques. In particular, we examined the interaction with ds DNA for the self-complementary strand 5′-[CAATCGGATCGAATTCGATCCGATTG]-3′, with the hybrid and basket G4 of hTel22 as well as the parallel G4 of Pu22 as the model of the c-myc oncogene. The photophysical behaviour of the complexes strongly depends on the type of DNA. Binding has been studied titrating 1 with different amounts of DNA monitoring absorption, fluorescence and circular dichroism (CD) spectra as well as the fluorescence lifetimes. We refer to ESI† for absorption and circular dichroism data. CD spectra (ESI,† Fig. S7) show that 1 binds to parallel G4 of Pu22, basket G4 and ds DNA not disturbing the conformation. Differently in the case of Tel22, we conclude that binding favours transition from the G4 hybrid structure to other G4 structures. Global analysis of the multiwavelength data set corresponding to the fluorescence spectra of the different mixtures in Fig. 2 allowed us to determine the best complexation model, the binding constants of the most stable complexes (Table 2) as well as the individual fluorescence spectra of the associated species (ESI,† Fig. S9).
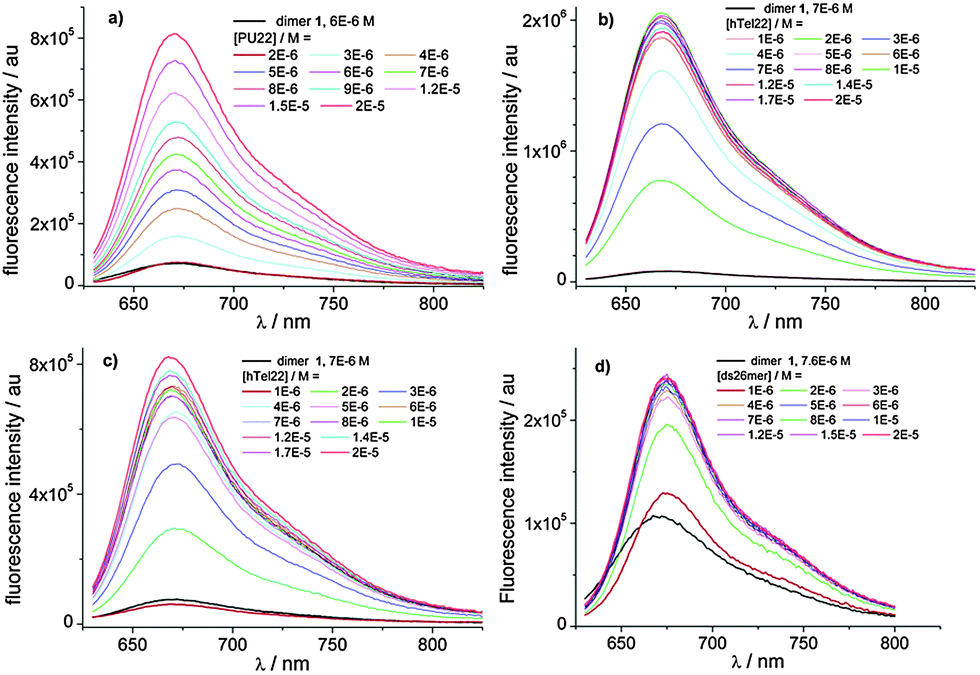 | ||
| Fig. 2 Fluorescence spectra of 7.0 × 10−6 M solutions of 1 with increasing DNA concentration (range 1 × 10−6–2.0 × 10−5 M) in phospate/KCl or NaCl buffer, pH 7.0. The spectrum of dimer 1 solution is the black line. (a) Pu22 with KCl; (b) hTel22 with KCl; (c) hTel22 with NaCl; and (d) ds26mer. | ||
| DNA | Stoichiometry DNA:ligand |
pK11 (M−1)/pK12a (M−2) | Φ F |
|---|---|---|---|
| a K 1i binding constant, obtained using the commercially available program Reactlab Equilibria. b Fluorescence quantum yield of the complex calculated using the spectra shown in ESI Fig. S9. | |||
| Pu22/KCl | 1:1 |
5.89 | 0.042 |
| 1:2 |
12.66 | — | |
| hTel22/KCl | 1:2 |
11.65 | 0.044 |
| hTel22/NaCl | 1:2 |
11.32 | 0.01 |
| ds26mer | 1:2 |
12.75 | 0.002 |
In the case of Pu22 the complexation model consists in the existence of two complexed species with 1:1 and 2:1 stoichiometry, only the 1:1 complex being fluorescent, while in the case of hTel22 with K+ and Na+ and ds26mer the analysis converged only with a complexation model of one fluorescent complex with 2:1 stoichiometry. Noticeably, we observed a 40-fold increase of the fluorescence quantum yield for the 1:1 complex of Pu22 and the 2:1 complex of htel22 with K+ compared to the isolated dimer (Table 2, ESI,† Fig. S9).
The selective turn-on effect upon complexation to G4 DNA by 1 (Fig. 3) is important from the point of view of possible applications of these molecules. 1a exhibits a less remarkable and selective turn-on emission upon binding, and for this reason, it has not been reported here (ESI,† Fig. S10).
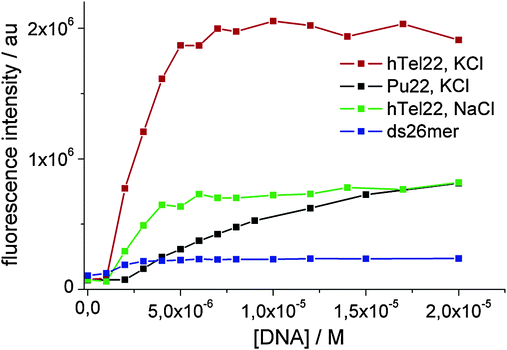 | ||
| Fig. 3 Fluorescence intensity enhancement of a 7 μM solution of 1 measured at 680 nm against DNA concentration. λexc. = 637 nm. | ||
Moreover, global analysis of the fluorescence decay data of 1 alone and in the presence of different concentrations of DNA obtained for excitation at 637 nm evidenced a different behaviour for the ds26mer NDI complexes. Only in the latter case, a tri-exponential function allowed convergence of global analysis while for G4 complexes a 4-exponential function was needed. In all fluorescent G4 complexes we observed a species with a long lifetime of ca. 5 ns (ESI,† Table S2) gaining importance with increasing DNA concentration, which is absent in the ds26mer complex (Fig. 4a). Fig. 4 also shows a graph with the average lifetime of a solution containing only NDI complexes.
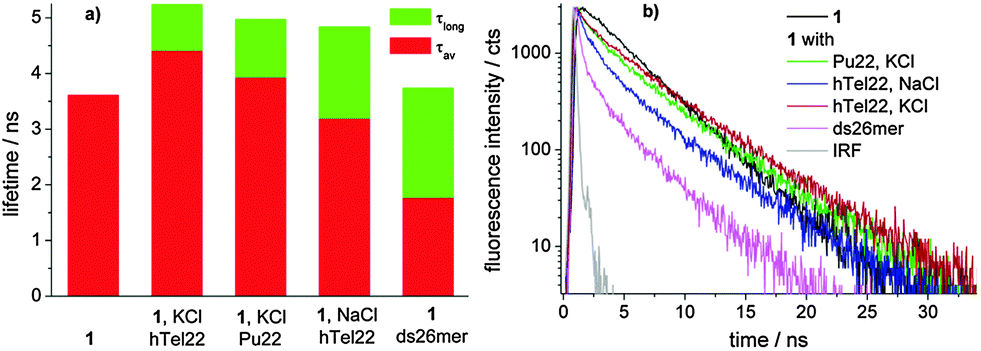 | ||
| Fig. 4 (a) Graph displaying the long lifetime component (τlong, in green) as well as the average lifetime (τav, in red) of solutions containing 7 × 10−6 M 1 and 2 × 10−5 M DNA in 0.01 M phosphate buffer of pH 7.0 with 100 mM KCl or NaCl. (b) Fluorescence decay of compound 1 alone and in the presence of excess DNA. | ||
The average lifetime (τav) of the ds DNA complex clearly differentiates from the average lifetime of the G4 complexes and this represents a very interesting tool to distinguish ds DNA NDI complexes from G4 NDI complexes. In all cases, the weak fluorescence ascribed to the tri-substituted NDI unit (red moiety in 1, Scheme 1) is completely quenched upon DNA complexation, paralleling the behaviour of NDI 2 upon hTel22 binding (ESI,† Fig. S11). Furthermore, the long fluorescence lifetime component of complex 1 obtained for excitation at 637 nm is similar to that of free 3 (4.4 ns)17 (Fig. 4). These data strongly suggest that the G4 binding moiety in dyad 1 is the tri-unit (red) and the flexible heptyl spacer allows the tetra-substituted (blue) moiety to assume a behaviour similar to that of free NDI 3. Indeed, the measured pKa values suggest that the protonation state of the two units has to be similar at pH 7. Electrostatic interactions of cationic G4 ligands with phosphate groups stabilizing the complexes are thus expected to be similar for both units. Other factors, such as steric hindrance and higher electron density on the aromatic core of the blue vs. red unit, may play a role in their different binding behaviour.
In conclusion, a water-soluble naphthalene diimide dyad conjugating red and blue NDIs was prepared and investigated as a fluorescent probe. The photophysical properties were thoroughly investigated by means of steady-state and time-resolved spectroscopy. Dyad 1 is a non-emitting molecule, unlike its NDI components. Upon complexation to G4 structures, the fluorescence of the dimer turns on in the red/NIR. Although the fluorescent probe does not exhibit remarkable selectivity between the investigated G4 structures, the G4 vs. ds selectivity is good. Furthermore, the average fluorescent lifetime of the G4 complexes with 1 is significantly different from the average fluorescent lifetimes of the ds complexes. This descriptor allows distinguishing the different types of complexes and it represents the most promising feature for the development of NDI dyads as fluorescent sensors for G4 structures by time-resolved fluorescence spectroscopy.
The Italian Ministry of Education, University and Research (MIUR), Rome (FIRB-Ideas RBID082ATK_003) and the Italian Association for Cancer Research (AIRC, IG2013-14708) financially supported this work.
Notes and references
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331 RSC
.
- G. W. Collie, R. Promontorio, S. M. Hampel, M. Micco, S. Neidle and G. N. Parkinson, J. Am. Chem. Soc., 2012, 134, 2723 CrossRef CAS PubMed
- F. Cuenca, O. Greciano, M. Gunaratnam, S. Haider, D. Munnur, R. Nanjunda, W. D. Wilson and S. Neidle, Bioorg. Med. Chem. Lett., 2008, 18, 1668 CrossRef CAS PubMed
- M. Micco, G. W. Collie, A. G. Dale, S. A. Ohnmacht, I. Pazitna, M. Gunaratnam, A. P. Reszka and S. Neidle, J. Med. Chem., 2013, 56, 2959 CrossRef CAS PubMed
- M. Di Antonio, F. Doria, S. N. Richter, C. Bertipaglia, M. Mella, C. Sissi, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2009, 131, 13132 CrossRef CAS PubMed
- F. Doria, M. Nadai, M. Folini, M. Di Antonio, L. Germani, C. Percivalle, C. Sissi, N. Zaffaroni, S. Alcaro, A. Artese, S. N. Richter and M. Freccero, Org. Biomol. Chem., 2012, 10, 2798 CAS
- F. Doria, M. Nadai, M. Folini, M. Scalabrin, L. Germani, G. Sattin, M. Mella, M. Palumbo, N. Zaffaroni, D. Fabris, M. Freccero and S. N. Richter, Chem. – Eur. J., 2013, 19, 78 CrossRef CAS PubMed
- M. Nadai, F. Doria, L. Germani, S. N. Richter and M. Freccero, Chem. – Eur. J., 2015, 21, 2330 CrossRef CAS PubMed
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593 CrossRef CAS PubMed
-
(a) J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908 CrossRef CAS PubMed
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406 CrossRef CAS PubMed
- A. Rizzo, E. Salvati, M. Porru, C. D'Angelo, M. F. Stevens, M. D'Incalci, C. Leonetti, E. Gilson, G. Zupi and A. Biroccio, Nucleic Acids Res., 2009, 37, 5353 CrossRef CAS PubMed
-
(a) F. Doria, M. Folini, V. Grande, G. Cimino-Reale, N. Zaffaroni and M. Freccero, Org. Biomol. Chem., 2015, 13, 570 RSC
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070 CrossRef PubMed
- F. Würthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem. – Eur. J., 2002, 8, 4742 CrossRef
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC
- F. Doria, I. Manet, V. Grande, S. Monti and M. Freccero, J. Org. Chem., 2013, 78, 8065 CrossRef CAS PubMed
- K. W. Zheng, D. Zhang, L. X. Zhang, Y. H. Hao, X. Zhou and Z. Tan, J. Am. Chem. Soc., 2011, 133, 1475 CrossRef CAS PubMed
-
(a) A. Renaud de la Faverie, A. Guedin, A. Bedrat, L. A. Yatsunyk and J.-L. Mergny, Nucleic Acids Res., 2014, 42, e65 CrossRef CAS PubMed
-
(a) J. Alzeer, P. J. C. Roth and N. W. Luedtke, Chem. Commun., 2009, 1970 RSC
- F. Doria, M. Nadai, G. Sattin, L. Pasotti, S. N. Richter and M. Freccero, Org. Biomol. Chem., 2012, 10, 3830 CAS
- J. E. Rogers, S. J. Weiss and L. A. Kelly, J. Am. Chem. Soc., 2000, 122, 427 CrossRef CAS
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Furstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Roger, F. Wurthner, N. Sakai and S. Matile, Science, 2006, 313, 84 CrossRef CAS PubMed
- Exciting 1 with SDS at 373 nm and measuring emission at 690 nm we observe a monoexponential decay indicating that only one species emits at 690 nm, so in the presence of SDS the tri-unit does not contribute at this wavelength, ESI,† Fig. S6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc01536g |
| This journal is © The Royal Society of Chemistry 2015 |