
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed borylation of arenes and indoles via C–H bond cleavage†
Takayuki
Furukawa
a,
Mamoru
Tobisu
*abc and
Naoto
Chatani
*a
aDepartment of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osakau.ac.jp; chatani@chem.eng.osakau.ac.jp
bCenter for Atomic and Molecular Technologies, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
cESICB, Kyoto University, Katsura, Kyoto 615-8510, Japan
First published on 11th March 2015
Abstract
The first nickel-catalyzed method for the borylation of carbon–hydrogen bonds in arenes and indoles is described. The use of an N-heterocyclic carbene ligand is essential for an efficient reaction, with an N-cyclohexyl-substituted derivative being optimal. This method is readily applied to the gram scale synthesis of 2-borylindole.
Over the past few decades, substantial progress has been made in the development of catalytic methods for C–H bond functionalization.1 Catalytic borylation of arenes is arguably the method of paramount importance, since it allows the introduction of a synthetically useful boron functionality directly at the C–H bond of an arene without using any directing groups.2 The regioselectivity of this method is predictable based on steric factors, enabling the widespread use of this borylation method in the late-stage elaboration of a range of complex aromatic molecules. Iridium complexes ligated with a bipyridine-based ligand are presently the catalyst of choice for the efficient, regioselective borylation of arenes (Scheme 1a).3 One remaining challenge in the catalytic C–H borylation methodology is the development of base metal catalysts, driven by recent concerns regarding the limited availability of precious metals. Although a seminal work by Hartwig demonstrated the potential activity of base metal complexes such as Fe and Mn in the C–H bond borylation reaction in 1995,4 catalytic variants were achieved only recently (Scheme 1b). Ohki and Tatsumi reported an iron-catalyzed borylation of furans and thiophenes in the presence of a hydrogen acceptor,5 while Kuang and Wang demonstrated that Fe2O3 nanoparticles can also catalyze the borylation of arenes in the presence of a more than stoichiometric amount of tert-butyl peroxide.6 A mechanistically unique C–H borylation using a Cu/Fe bimetallic catalyst under photochemical conditions was developed by Mankad.7 Chirik reported a cobalt-catalyzed C–H borylation applicable to a broad range of arenes and heteroarenes.8 Despite these advances, the full potential of base metals to promote C–H borylation in a broader sense, to promote C–H activation, has not been explored, especially in the case of metals other than iron9 and cobalt.10 Herein, we report the first nickel-catalyzed borylation of C–H bonds in arenes and indoles.
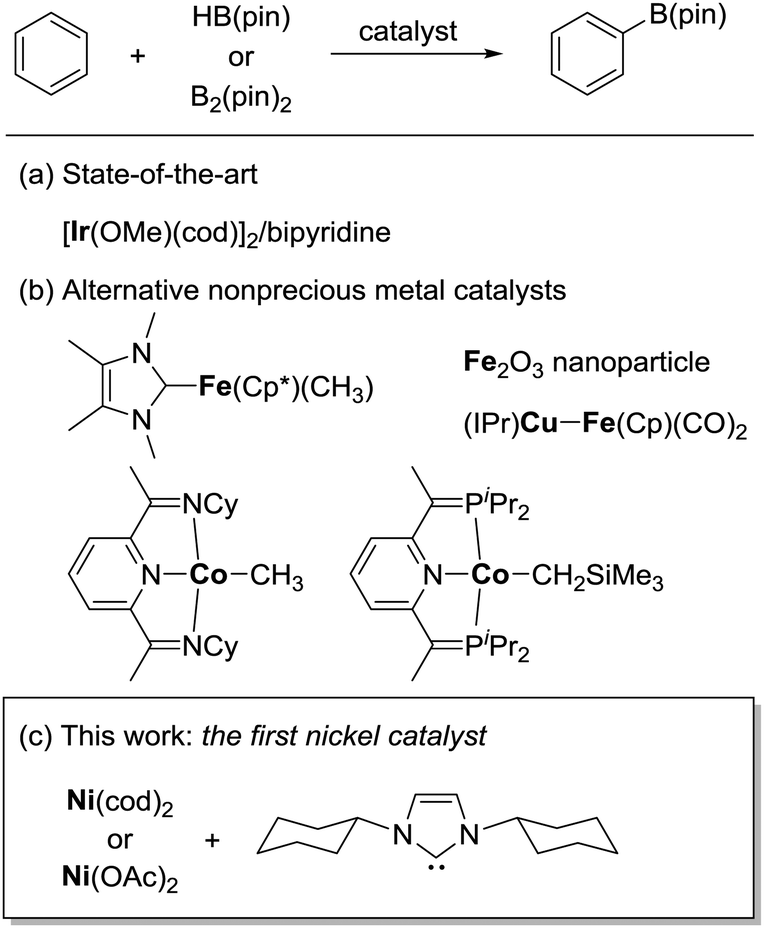 | ||
| Scheme 1 Catalysts for the borylation of C–H bonds of (hetero)arenes. | ||
During the course of our mechanistic studies of the Ni(cod)2/PCy3-catalyzed reductive cleavage of anilides using HBpin, we found that the hydrogens of the aromatic ring of the toluene solvent were interchanged with those of the HBpin.11 This observation clearly indicated that the C–H bonds in simple arenes can be activated under nickel catalysis. We were intrigued by this finding because, to the best of our knowledge, the nickel-catalyzed C–H functionalization of simple arenes (i.e., arenes bearing no directing group) has not yet been reported, with the exception of the reactions using highly fluorinated benzenes.12,13 We therefore envisaged that the C–H borylation of simple arenes by nickel should be possible if an appropriate catalyst system can be identified.
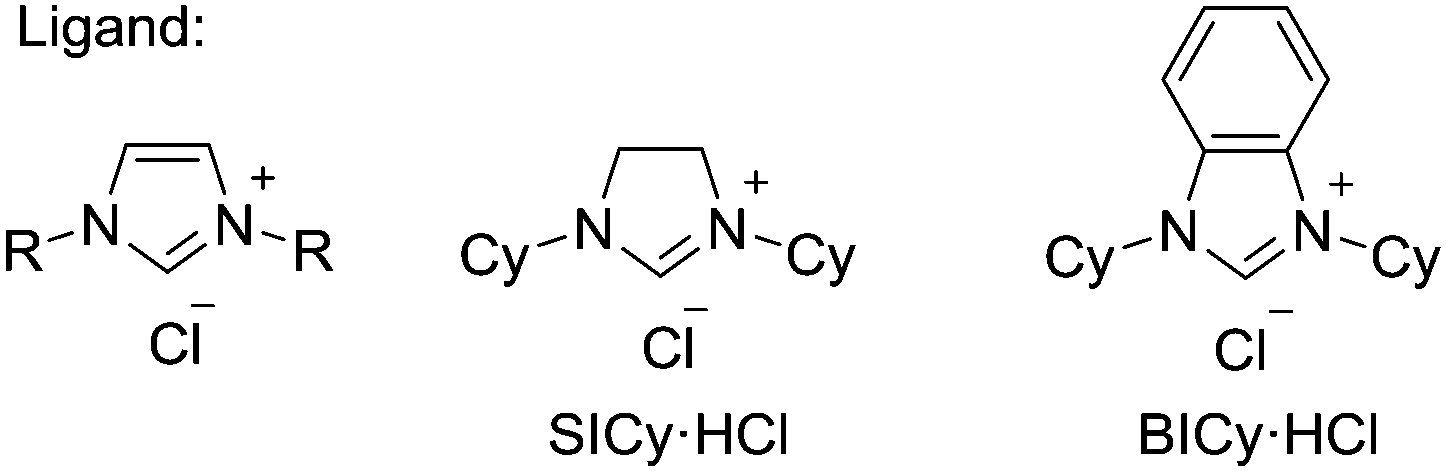
We initially examined the nickel-catalyzed reaction of HBpin with benzene in the presence of a variety of ligands (Table 1). Although we previously determined that the Ni(cod)2/PCy3 system can mediate the H/D exchange reaction via C–H activation of arenes,11 this catalyst system did not lead to the formation of the desired borylated product 1 (entry 2). In contrast, the use of IMes, an N-heterocyclic carbene (NHC) bearing a mesityl group, was found to generate 1 in 51% yield (entry 3). Replacing the mesityl group with either the larger 2,6-iPr2C6H2 group (i.e., IPr) or the smaller methyl group (i.e., IMe) decreased the yield of 1 (entries 4 and 5). Further experimentation with the substituent on the nitrogen of the NHC ligand revealed that the cyclohexyl-substituted derivative (i.e., ICy) was the most effective ligand among those tested, with 1 being formed in 71% isolated yield (entry 8). Whereas the use of a saturated analog of ICy (i.e., SICy) led to a complete loss of catalytic activity (entry 11), the benzohomolog of ICy (i.e., BICy) promoted the borylation of benzene with comparable efficiency (entry 12).
|
|
||
|---|---|---|
| Entry | Ligand | Yieldb (%) |
| a Reaction conditions: HBpin (1.2 mmol), Ni(cod)2 (0.036 mmol), ligand (0.036 mmol), NaOtBu (0.072 mmol) and benzene (1.0 mL) in a screw-capped vial under N2 at 80 °C for 20 h. b GC yield based on HBpin. c NaOtBu was not added. d Isolated yield. | ||
| 1 | None | 5 |
| 2c | PCy3 | 0 |
| 3 | IMes·HCl (R = 2,4,6-Me3C6H2) | 51 |
| 4 | IPr·HCl (2,6-iPr2C6H3) | 16 |
| 5 | IMe·HCl (R = Me) | 22 |
| 6 | IiPr·HBF4 (R = iPr) | 33 |
| 7 | ItBu·HCl (R = tBu) | 50 |
| 8 | ICy·HCl (R = cyclohexyl) | 75 (71)d |
| 9 | I(1-Ad)·HCl (R = 1-adamantyl) | 36 |
| 10 | I(2-Ad)·HCl (R = 2-adamantyl) | Trace |
| 11 | SICy·HCl | 2 |
| 12 | BICy·HCl | 69 |
|
||
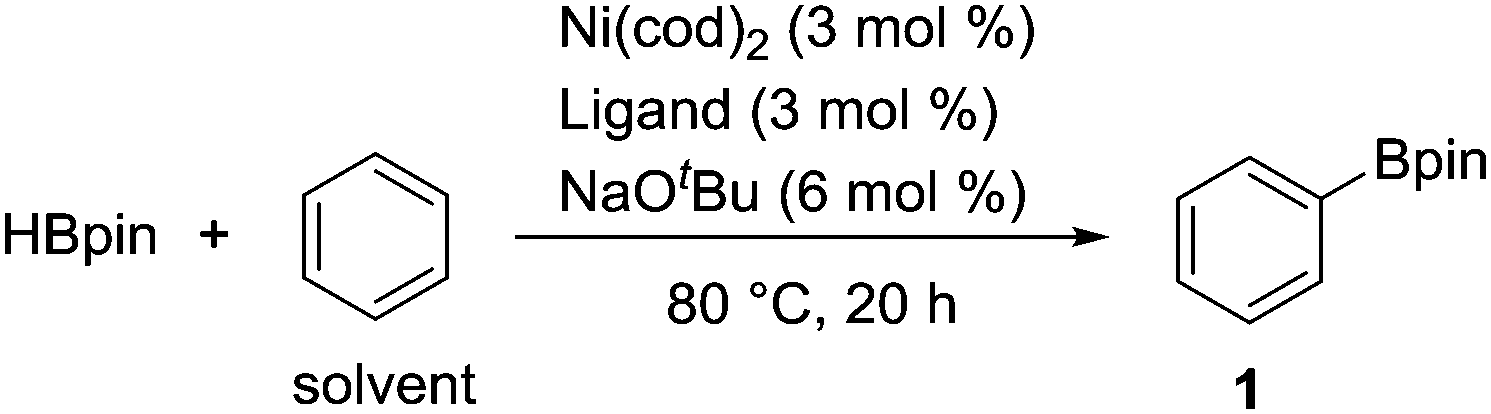
B2pin2 was also found to be a suitable boron source for this nickel-catalyzed borylation, forming 1 in a 125% yield based on B2pin2 (eqn (1)).
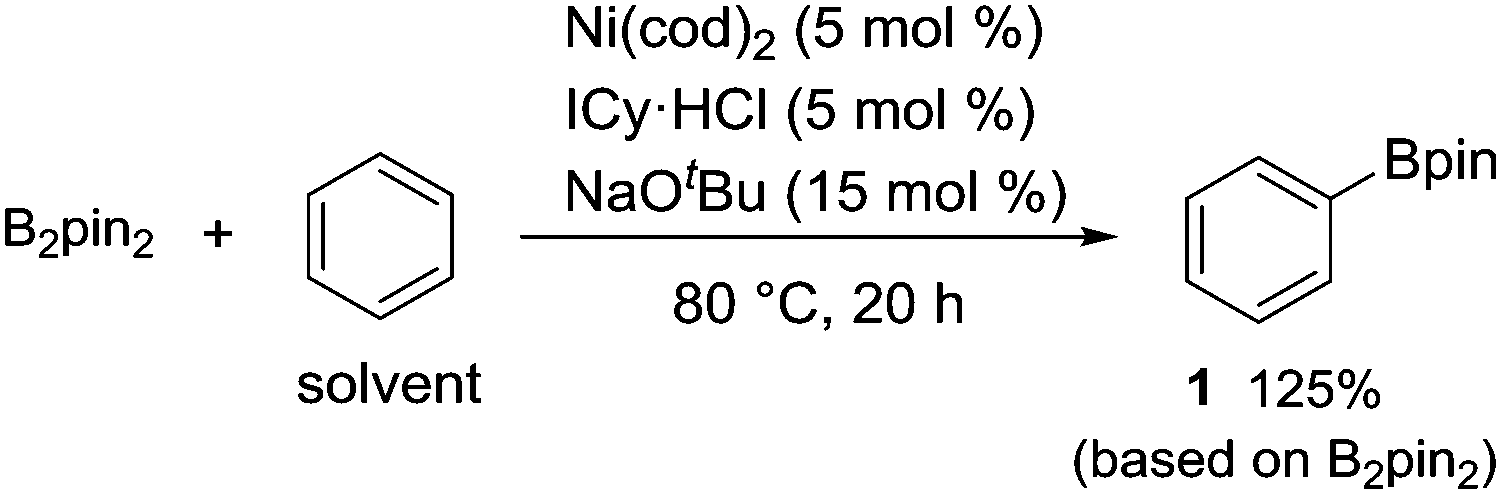 | (1) |
|
|
||
|---|---|---|
| Entry | Arene | Productb |
| a Reaction conditions: HBpin (1.2 mmol), Ni(cod)2 (0.035 mmol), ICy·HCl (0.035 mmol), NaOtBu (0.070 mmol) and arene (1.0 mL) in a screw-capped vial under N2 at 100 °C for 20 h. b Isolated yield based on HBpin. c At 80 °C. d N-Methylpyrrole (2.0 mmol) in methylcyclohexane (1.0 mL). | ||
| 1 |
|
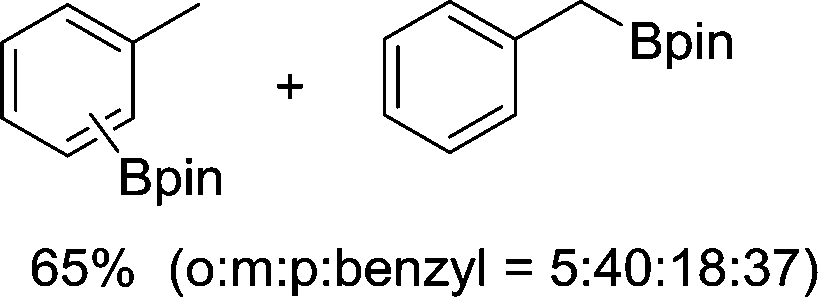
|
| 2 |
|
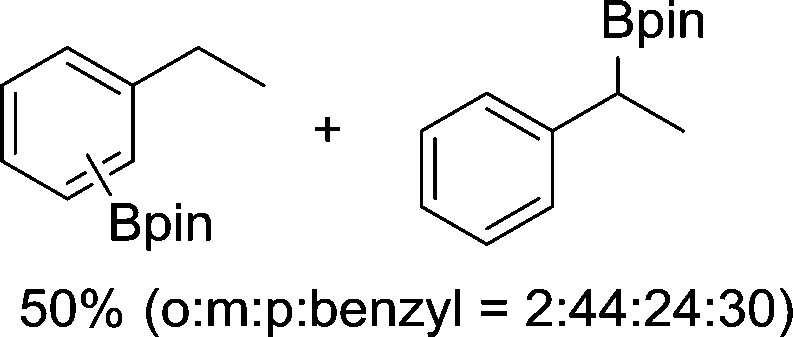
|
| 3 |
|
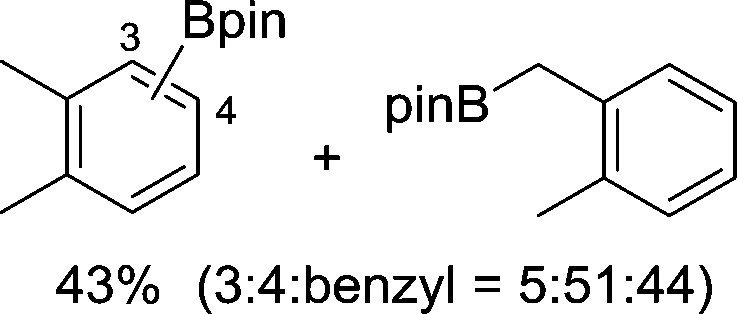
|
| 4 |
|
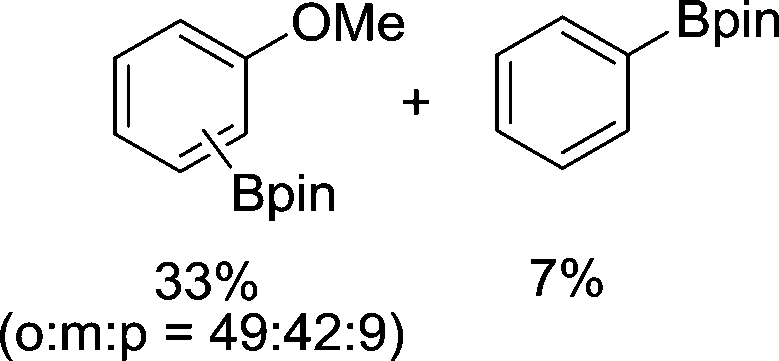
|
| 5c |
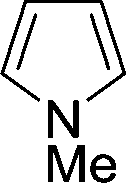
|
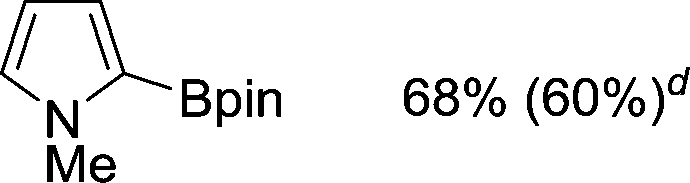
|
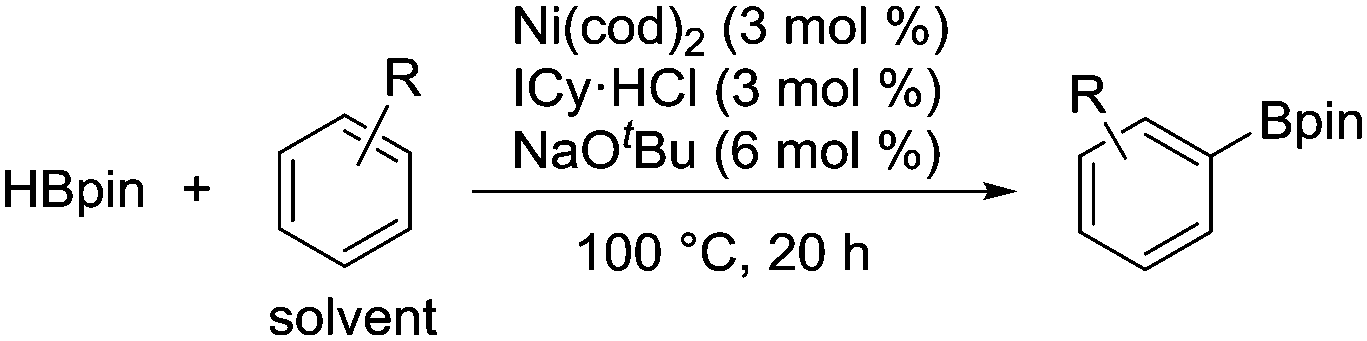
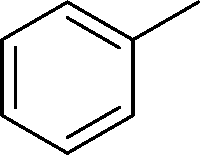
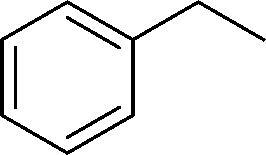
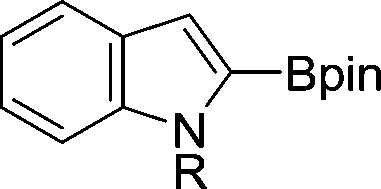
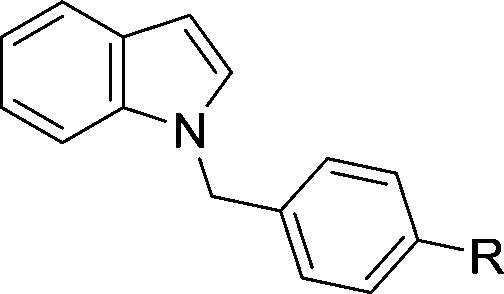
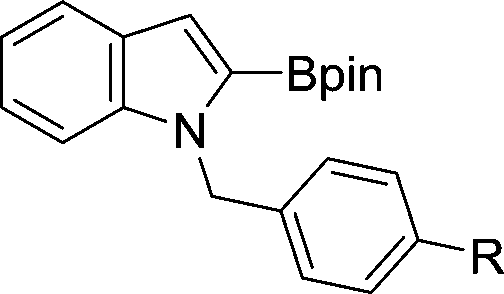
The evident high reactivity of pyrrole prompted us to examine indole derivatives in this nickel-catalyzed borylation reaction (Table 3). As expected, the borylation occurred efficiently using indole as the limiting agent and also took place regioselectively at the 2-position. In addition to N-methylindole (entry 1), a bulkier N-butyl-substituted indole successfully underwent borylation without any decrease in the regioselectivity of the reaction (entry 2). An N,O-acetal moiety remained intact, serving as a suitable protecting group in this reaction (entry 3). In addition, N-benzyl groups were also tolerated, leaving the aryl group free from borylation (entries 4 and 5). Installation of a boryl group at the sterically hindered 2-position of 3-substituted indoles was also possible (entry 6). The reactivity and selectivity of this borylation was unaffected by the introduction of a methoxy group to the indole framework (entry 7). Although a C(aryl)–F bond can be activated by a low valent nickel catalyst,17 fluoroindole underwent borylation with its fluoride moiety remaining intact (entry 8). Azaindole also served as a good substrate for this borylation, generating the corresponding 2-borylated product (entry 9).
|
|
|||
|---|---|---|---|
| Entry | Indole | Product | Isolated yield (%) |
| a Reaction conditions: indole (0.80 mmol), HBpin (1.2 mmol), Ni(cod)2 (0.040 mmol), ICy·HCl (0.040 mmol), NaOtBu (0.080 mmol), and methylcyclohexane (1.0 mL) in a screw-capped vial under N2 at 80 °C for 16 h. b HBpin (1.2 equiv.) was used. | |||
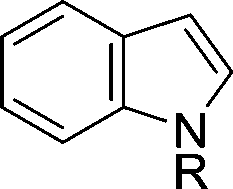
|
|
||
| 1 | R = Me | 78 | |
| 2 | R = n-butyl | 68 | |
| 3 | R = CH2OMe | 68 | |
|
|
||
| 4 | R = H | 82 | |
| 5 | R = OMe | 76 | |
| 6 |
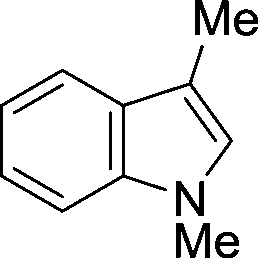
|
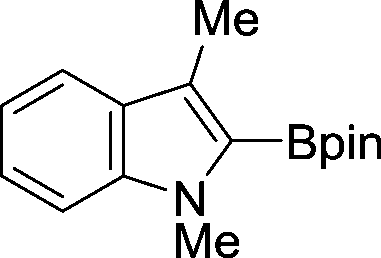
|
87 |
| 7 |
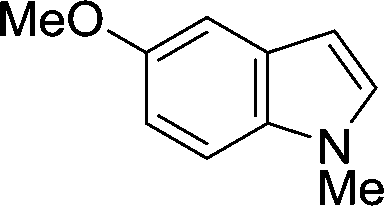
|
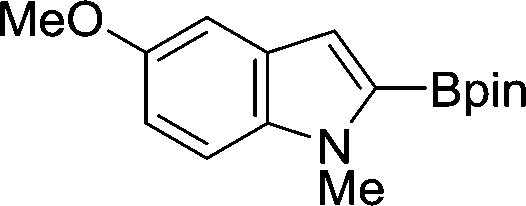
|
76 |
| 8b |
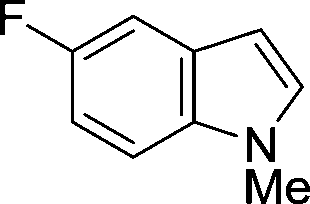
|
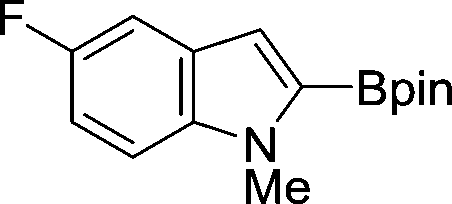
|
82 |
| 9b |
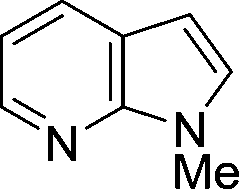
|
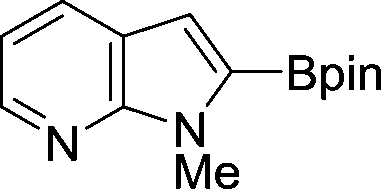
|
56 |
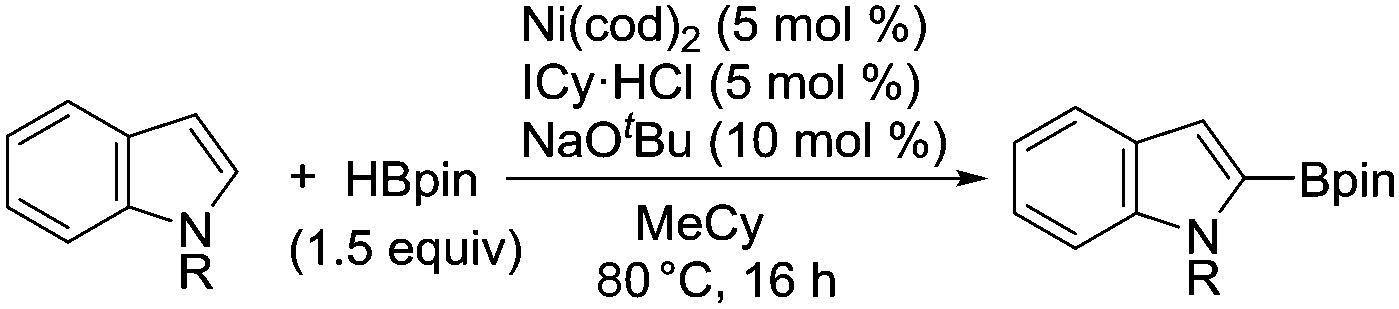
Although Ni(cod)2 was routinely used as the catalyst for this borylation during our exploratory studies, Ni(OAc)2 was found to serve as a more useful catalyst precursor. A protocol using Ni(OAc)2 allowed the successful implementation of this borylation on the gram-scale, highlighting the practical utility of the nickel-catalyzed system (Scheme 2).
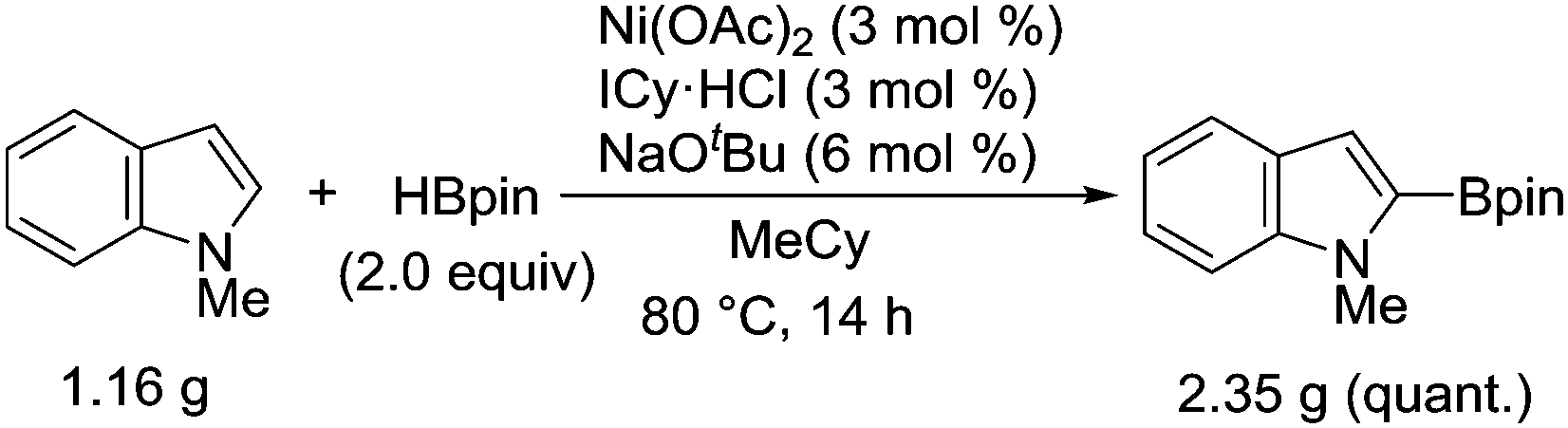 | ||
| Scheme 2 Gram scale synthesis of 2-borylindole using Ni(OAc)2. | ||
To obtain further insights into the mechanistic aspects of the nickel-catalyzed borylation, some preliminary mechanistic experiments were performed. Comparison of the initial rates of the nickel-catalyzed borylation reactions of benzene and benzene-d6 determined that kH/kD was 2.1, which was slightly smaller than the values reported for iridium2 and iron7 systems. The nickel-catalyzed borylation of benzene or indole was also found to be completely inhibited when the reaction was attempted in the presence of an excess of mercury. In addition, filtration tests indicated that the liquid phase of the catalytic mixture did not contain catalytically active species (see ESI† for details). The currently available data indicate that the borylation is likely mediated by heterogeneous nickel species, although more detailed studies are required for a definitive understanding of the nature of the catalysis.18 Finally, a recent report on the KOtBu-catalyzed silylation of heteroarenes19,20 motivated us to examine the borylation of indole in the presence of only NaOtBu. The borylation product was not observed under such conditions, indicating that the nickel species play an essential role in this borylation reaction.
In summary, we have shown for the first time that the direct borylation of arenes can be mediated by a nickel-based catalyst. Indoles are particularly reactive with this catalyst system, affording the corresponding 2-borylated products. The successful use of nickel is noteworthy because nickel is rarely employed as a catalyst for the C–H bond activation process in the absence of a directing group.12 The heterogeneous nature of the active species generated under the current conditions could be important in explaining the unprecedented reactivity of nickel,21 although further investigations are required for a complete understanding of the mechanism of this borylation. The application of these nickel species to other transformations as well as catalytic borylations using other base metals are currently being investigated in our laboratories.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” from MEXT, Japan. M.T. was also supported by the “Elements Strategy Initiative to Form Core Research Center” from MEXT. T.F. expresses his special thanks for a JSPS Research Fellowship for Young Scientists. We also thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for assistance with the HRMS analyses.
Notes and references
- Selected recent reviews: (a) F. Kakiuchi, T. Kochi and S. Murai, Synlett, 2014, 2390 CrossRef CAS PubMed; (b) M. Miura, T. Satoh and K. Hirano, Bull. Chem. Soc. Jpn., 2014, 87, 751 CrossRef CAS; (c) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (f) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (g) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (h) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (i) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (j) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (k) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- A review: I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- A seminal work: T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed.
- K. M. Waltz, X. He, C. Muhoro and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11357 CrossRef CAS.
- T. Hatanaka, Y. Ohki and K. Tatsumi, Chem. – Asian J., 2010, 5, 1657 CrossRef CAS PubMed.
- G. Yan, Y. Jiang, C. Kuang, S. Wang, H. Liu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 3170 RSC.
- T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- Selected reviews on C–H bond functionalization under Fe catalysis: (a) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (b) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (c) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed.
- Selected reviews on C–H bond functionalization under Co catalysis: (a) N. Yoshikai, Bull. Chem. Soc. Jpn., 2014, 87, 843 CrossRef CAS; (b) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (c) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed.
- M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587 CrossRef CAS PubMed A related work using a nickel cluster: R. Beck, M. Shoshani and S. A. Johnson, Angew. Chem., Int. Ed., 2012, 51, 11753 CrossRef PubMed.
- (a) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 16170 CrossRef CAS PubMed; (b) K. S. Kanyiva, N. Kashihara, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, Dalton Trans., 2010, 39, 10483 RSC; (c) M. E. Doster, J. A. Hatnean, T. Jeftic, S. Modi and S. A. Johnson, J. Am. Chem. Soc., 2010, 132, 11923 CrossRef CAS PubMed; (d) M. E. Doster and S. A. Johnson, Organometallics, 2013, 32, 4174 CrossRef CAS; (e) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed.
- Nickel-catalyzed C–H functionalization of arenes bearing a directing group: L. C. M. Castro and N. Chatani, Chem. Lett. DOI:10.1246/cl.150024.
- S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS.
- Borylation of benzylic C–H bond by Pd/C catalyst: T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 1082 CrossRef CAS.
- Reviews on nickel-catalyzed transformations via C–OMe cleavage of aryl ethers: J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC.
- Selected works on C(aryl)–F bond activation under Ni catalysis: (a) Y. Kiso, K. Tamao and M. Kumada, J. Organomet. Chem., 1973, 50, C12 CrossRef CAS; (b) B.-J. Li, Y.-Z. Li, X.-Y. Lu, J. Liu, B.-T. Guan and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 10124 CrossRef CAS PubMed; (c) N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590 CrossRef CAS PubMed; (d) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505 CrossRef CAS PubMed; (e) A. D. Sun, K. Leung, A. D. Restivo, N. A. LaBerge, H. Takasaki and J. A. Love, Chem. – Eur. J., 2014, 20, 3162 CrossRef CAS PubMed; (f) H. Saijo, H. Sakaguchi, M. Ohashi and S. Ogoshi, Organometallics, 2014, 33, 3669 CrossRef CAS; (g) M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80 CrossRef CAS PubMed.
- A related base effect in Ir-catalyzed C–H borylation: (a) M. N. Eliseeva and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15169 CrossRef CAS PubMed; (b) T. Ohmura, T. Torigoe and M. Suginome, Chem. Commun., 2014, 50, 6333 RSC.
- Unique reactivity of heterogeneous nickel species in C–O bond activation: A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of new products. See DOI: 10.1039/c5cc01378j |
| This journal is © The Royal Society of Chemistry 2015 |