
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combination of automated solid-phase and enzymatic oligosaccharide synthesis provides access to α(2,3)-sialylated glycans†
Richard J.
Fair
a,
Heung Sik
Hahm
ab and
Peter H.
Seeberger
*ab
aDepartment of Biomolecular Systems, Max-Planck-Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: peter.seeberger@mpikg.mpg.de; Fax: +49 30 838-59302; Tel: +49 30 838-59301
bFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany
First published on 2nd March 2015
Abstract
A synthetic strategy combining automated solid-phase chemical synthesis and enzymatic sialylation was developed to access α(2,3)-sialylated glycans.
N-Acetyl-neuraminic acid (Neu5Ac) is the most studied among the over 50 sialic acids described to date and is a component of many glycans of biological relevance. α(2,6)-Sialylated and α(2,3)-silylated glycans are predominantly found in nature.1–3 Sialylated glycans on the cell surface regulate various cellular functions4 and are linked to a host of extracellular recognition events.5,6 Sialic acid containing glycans are associated with many cancers,7–9 autoimmune disorders,10 and diabetes11 but are also exploited by pathogenic organisms including trypanosomes,12 bacteria,13 and viruses.14 Biological studies greatly benefit from the availability of defined glycans as molecular probes.
Sialylated oligosaccharides have been a challenge for synthetic chemists since no participating group can be placed at the neighboring C-3 position and next to the sterically hindered, quaternary anomeric center is an electron withdrawing carbonyl that diminishes the reactivity. Significant progress has been made in developing efficient chemical sialylation reactions but glycosylation efficiencies for these linkages still remain significantly lower than those obtained for other couplings.2,15,16 The automated assembly of glycans on solid support benefits from mass action of the glycosylating agent and has resulted in the rapid assembly of ever more complex glycans.16,17 However, the automated assembly of sialylated glycans has been challenging. Sialic acid-galactose disaccharide building blocks were used to produce α(2,3)- and α(2,6)-sialylated glycans by automated solid-phase synthesis.18 The use of disaccharide building blocks is not ideal within the logic framework of glycan assembly based on monosaccharide building blocks. More recently, a sialic acid monosaccharide building block proved useful in producing α(2,6)-sialylated glycans.19 α(2,3)-Silylated glycans were formed in low yields (<20%) using this method.
To address the challenge that could not be met with chemical means alone, the combination of automated glycan assembly and enzymatic glycan synthesis, two complementary techniques, might offer a solution. Here, we demonstrate that glycans assembled by automated synthesis can be used as scaffolds for enzymatic elaboration to access the desired sialylated glycans. Sialyltransferases transfer sialic acids from activated cytodine monophosphate (CMP)-sialic acids to oligosaccharide substrates.1 Bacterial sialyltransferases have been reliable tools to efficiently and selectively install desired sialic acids2,20 even on gram scale.21 By combining the power of automated glycan assembly to prepare the glycan backbone with the efficiency and selectivity of glycan sialylation, this strategy combines the advantages inherent to both methods for the rapid production sialylated glycans.
Five oligosaccahrides GM3 (1), GM1b (cisGM1, 2), sialyl LC4 (3), SPG (sialyl nLC4, 4), and SLPG (nHM1, 5) (Fig. 1) were selected to explore the combination of automated glycan assembly and enzymatic sialylation. These sialosides are structurally similar as they have linear, non-fucosylated backbones and terminal sialic acids since the sialyltransferase used for these proof-of-principle studies, a truncated Pasturella multocida sialyltransferase 1 (PmST1), operates on such linear structures.
 | ||
| Fig. 1 α(2,3)-Sialic acid containing target glycans. | ||
α(2,3)-Sialyltransferases glycosylate terminal galactose residues and moieties immediately next to the galactose effect the sialylation efficiency.22 To evaluate the influence of the nearest neighbor of galactose on the enzymatic reactions, different sugars and linkages next to the galactose residue were explored. The target molecules are either tumor-associated carbohydrate antigens (TACAs) (1, 2, 4),7,23–25 associated with autoimmune disorders (1, 2, 4, 5),10,26–28 or exploited by pathogens (1, 4).13,29,30
The synthetic strategy, exemplified for the synthesis of GM1b (2) (Scheme 1) relies on the automated solid phase assembly of the glycan backbone (11) using thioglycoside building blocks (6–9) and resin appended with a photo-cleavable linker (10)31 through successive cycles of glycosylation. All reaction steps including removal of the fluorenylmethloxycarbonyl (Fmoc) temporary protecting group and acidic washes are carried out on an automated synthesizer.32 Building blocks were activated using N-iodosuccinimide (NIS) and triflic acid (TfOH) and two couplings using each 4.4 equivalents of building block in all cases except for SLPG (5) where five equivalents of building block per cycle were used. Building blocks (6, 8, 9, S3–S5, ESI†) have been developed previously33–35 and were known to couple efficiently and selectively. Fmoc protecting groups were removed by exposure to a solution of triethylamine (TEA) (20% in dimethylformamide (DMF)). An acid wash with trimethylsilyltriflate (TMSOTf) follows to ensure the removal of base prior to the introduction of the next building block. UV irradiation of the resin in a continuous flow device afforded fully protected oligosaccharides.31 Yields for protected glycosides were high with only very small quantities of deletion sequences observed by analytical HPLC. Overall yields following HPLC purification ranged from 36% to 46% with the number of automated steps ranging from four steps for the disaccharide precursor to GM3 to twelve steps for the hexasaccharide precursor of SLPG (Table 1).
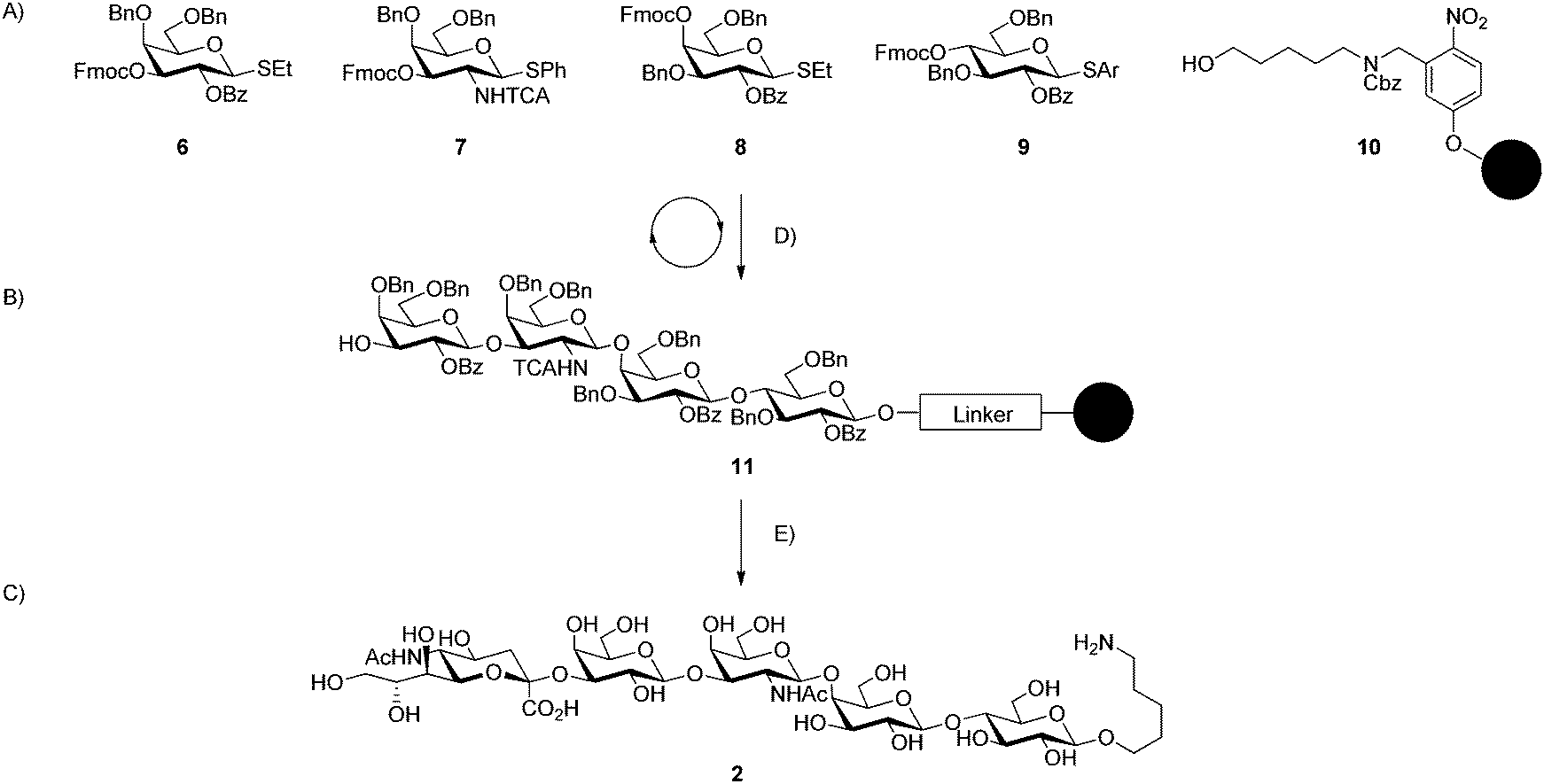 | ||
| Scheme 1 Automated glycan assembly synthetic strategy illustrated for GM1b (2). (A) Thioglycoside building blocks and photo-cleavable resin; (B) fully protected, resin-bound glycoside; (C) α(2,3)-sialylated glycoside; (D) automated steps: (1) glycosylation (NIS, TfOH, DCM, Dioxane), (2) Fmoc deprotection (TEA, DMF), (3) acidic wash (TMSOTf, DCM); (E) post-automation steps: (1) photo-cleavage (hν, DCM), (2) methanolysis (NaOMe, MeOH, DCM), (3) hydrogenolysis (Pd/C, H2, MeOH, H2O, AcOH), (4) sialylation (α(2,3)-sialyltransferase, CMP-Neu5Ac, alkaline phosphatase, Tris-HCl buffer). | ||
| Target glycan | Automation (overall)b | Global deprotectionc | Sialylationd |
|---|---|---|---|
| a All yields given as % values. b Yield of protected glycan following photo-cleavage from resin and HPLC purification as determined based on resin loading. c Yield of deprotected glycan following methanolysis, hydrogenolysis, and HPLC purification. d Yield of enzymatic sialylation following HPLC purificaiton. | |||
| GM3 (1) | 42 | 82 | 87 |
| GM1b (2) | 46 | 91 | 78 |
| sLC4 (3) | 36 | 83 | 79 |
| SPG (4) | 44 | 86 | 87 |
| SLPG (5) | 37 | 78 | 89 |
All remaining protective groups were removed to obtain completely deprotected oligosaccharides. Benzoyl esters were removed by methanolysis using a solution of sodium methoxide (NaOMe) in methanol and dichloromethane (DCM) at 40 °C. Subsequent hydrogenolysis of benzyl ethers and the carboxybenzyl (Cbz) carbamate with concurrent reduction of trichloroacetyl (TCA) groups was achieved upon exposure to 45 psi hydrogen with a Pd/C catalyst in a methanol, water, and acetic acid solution. Following global deprotection, the products were HPLC purified to yield substrates for the α(2,3)-sialyltransferase. The deprotection steps yielded 78–91% of the desired glycans over two steps (Table 1). The free glycans are appended with an amine functionalized linker that serves as a convenient handle for ligation to microarrays or carrier proteins used in vaccine conjugates.
With several oligosaccharides in hand, a truncated α(2,3)-sialyltransferase from Pasturella multocida (PmST1) was employed for enzymatic sialylation. This commercially available enzyme has a broad substrate specificity and has been used successfully in enzymatic syntheses.22
The sialylation reactions employed α(2,3)-sialyltransferase, CMP-Neu5Ac, and an alkaline phosphatase that serves to dephosphorylate the CMP byproduct of the reaction. Reactions proceeded in a Tris-HCl buffer at pH 8.0 at 37 °C with high conversion (>95% as determined by analytical HPLC) for all substrates with 1,4-linked terminal galactose residues (GM3, SPG, and SLPG precursors) using 1.1 equivalents of CMP-Neu5Ac and 30 mU sialyltransferase per mmol substrate. Sialylation was less efficient for substrates with 1,3-linked galactoses (GM1b and sLC4 precursors), however. To achieve high conversion (86%) in the case of sLC4 more enzyme was necessary (120 mU sialyltransferase per mmol substrate). In the case of GM1b, high conversion (87%), required more enzyme and four equivalents of CMP-Neu5Ac. Isolated yields, following HPLC purification of the target sialosides ranged from 78–89% (Table 1).
Automated glycan assembly and enzymatic synthesis were combined to produce five α(2,3)-sialylated glycans. Solid-phase chemistry is used for the rapid and high yielding production of the glycan backbones using just a few building blocks, while the sialyltransferase is used for high yielding and highly regio- and stereoselective sialylation. The combination of automated and enzymatic glycosylations is expected to be of general utility. Future work will seek to apply this approach to other enzymes.
We thank the Max-Planck Society and the European Research Council (ERC Advanced Grant AUTOHEPARIN to PHS) for generous financial support.
Notes and references
- Essentials of Glycobiology, ed. A. Varki, R. Cummings, J. Esko, H. Freeze, G. Hart and J. Martht, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 2009 Search PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 164–176 Search PubMed.
- D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C.-W. von der Lieth and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 685–691 CrossRef CAS PubMed.
- K. Furukawa, K. Hamamura, W. Aixinjueluo and K. Furukawa, Ann. N. Y. Acad. Sci., 2006, 1086, 185–198 CrossRef CAS PubMed.
- I. Bucior and M. M. Burger, Curr. Opin. Struct. Biol., 2004, 14, 631–637 CrossRef CAS PubMed.
- R. Schauer, Curr. Opin. Struct. Biol., 2009, 19, 507–514 CrossRef CAS PubMed.
- S. I. Hakomori and Y. Zhang, Chem. Biol., 1997, 4, 97–104 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526–542 CrossRef CAS PubMed.
- R. A. C. Hughes and D. R. Cornblath, Lancet, 2005, 366, 1653–1666 CrossRef CAS.
- E. Cabrera-Rode, O. Diaz-Horta, L. E. Fernandez, A. Carr, G. Marquina, O. Valiente, R. M. Gonzalez-Suarez and A. Uriarte, Autoimmunity, 1995, 20, 145–151 CrossRef CAS PubMed.
- R. Schauer and J. P. Kamerling, ChemBioChem, 2011, 12, 2246–2264 CrossRef CAS PubMed.
- C. S. Berenson, K. B. Sayles, J. Huang, V. N. Reinhold, M. A. Garlipp and H. C. Yohe, FEMS Immunol. Med. Microbiol., 2005, 45, 171–182 CrossRef CAS PubMed.
- J. Fantini, D. Hammache, G. Pieroni and N. Yahi, Glycoconjugate J., 2000, 17, 199–204 CrossRef CAS PubMed.
- M. J. Kiefel and M. von Itzstein, Chem. Rev., 2002, 102, 471–490 CrossRef CAS PubMed.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS PubMed.
- P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 19–28 RSC.
- D. Esposito, M. Hurevich, B. Castagner, C.-C. Wang and P. H. Seeberger, Beilstein J. Org. Chem., 2012, 8, 1601–1609 CrossRef CAS PubMed.
- C.-H. Lai, H. S. Hahm, C.-F. Liang and P. H. Seeberger, submitted.
- R. M. Schmaltz, S. R. Hanson and C.-H. Wong, Chem. Rev., 2011, 111, 4259–4307 CrossRef CAS PubMed.
- S. Fort, L. Birikaki, M. P. Dubois, T. Antoine, E. Samain and H. Driguez, Chem. Commun., 2005, 2558–2560 RSC.
- H. Yu, H. Chokhawala, R. Karpel, H. Yu, B. Wu, J. Zhang, Y. Zhang, Q. Jia and X. Chen, J. Am. Chem. Soc., 2005, 127, 17618–17619 CrossRef CAS PubMed.
- M. H. Ravindranath, S. Muthugounder, N. Presser, S. R. Selvan, A. D. Santin, S. Bellone, T. S. Saravanan and D. L. Morton, Biochem. Biophys. Res. Commun., 2007, 353, 251–258 CrossRef CAS PubMed.
- Z. Wang, L. Wen, X. Ma, Z. Chen, Y. Yu, J. Zhu, Y. Wang, Z. Liu, H. Liu, D. Wu, D. Zhou and Y. Li, Glycobiology, 2012, 22, 930–938 CrossRef CAS PubMed.
- Y. Kawaguchi, Y. Miyamoto, T. Inoue and Y. Kaneda, Int. J. Cancer, 2009, 124, 2478–2487 CrossRef CAS PubMed.
- Y. Tsukuda, N. Iwasaki, N. Seito, M. Kanayama, N. Fujitani, Y. Shinohara, Y. Kasahara, T. Onodera, K. Suzuki, T. Asano, A. Minami and T. Yamashita, PLoS One, 2012, 7, e40136 CAS.
- A. Ikeda, A. Komatsuzaki, T. Kasama, S. Handa and T. Taki, Biochim. Biophys. Acta, 2000, 81–90 CrossRef CAS.
- N. Yuki, Y. Tagawa and S. Handa, J. Neuroimmunol., 1996, 70, 1–6 CrossRef CAS PubMed.
- S. S. Rawat, S. A. Gallo, J. Eaton, T. D. Martin, S. Ablan, V. N. Kewal-Ramani, J. M. Wang, R. Blumenthal and A. Puri, J. Virol., 2004, 78, 7360–7368 CrossRef CAS PubMed.
- G. Nyberg, N. Stromberg, A. Jonsson, K. A. Karlsson and S. Normark, Infect. Immun., 1990, 58, 2555–2563 CAS.
- S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5858–5861 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- H. S. Hahm, M. Schlegel, M. Hurevich, S. Eller and P. H. Seeberger, in preparation.
- O. Calin, S. Eller, H. S. Hahm and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 3995–4002 CrossRef CAS PubMed.
- L. Kröck, D. Esposito, B. Castagner, C. C. Wang, P. Bindschädler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra, and analytical HPLC traces. See DOI: 10.1039/c5cc01368b |
| This journal is © The Royal Society of Chemistry 2015 |