
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal assisted cyclodimerization of doubly N-confused dipyrrins into planar aza-heptalenes†
Santosh C.
Gadekar
,
Baddigam Kiran
Reddy
and
Venkataramanarao G.
Anand
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Pune – 411008, Maharashtra, India. E-mail: vg.anand@iiserpune.ac.in
First published on 17th April 2015
Abstract
Metal salts cyclodimerize doubly N-confused dipyrrin into a nornorrole type macrocycle, with a bipyrrolic unit at its center. It also represents an unusual aza-heptalene structure with fused bicyclic seven membered rings. The fused rings can have one or two C–N bonds between the dipyrrin units in the cyclodimer. The 1H NMR spectrum of these molecules displays aromatic character, rather than antiaromatic behaviour expected of nornorrole, and planar conformation in the solid state.
A dipyrrin can ligate a variety of metal ions to yield the corresponding metal chelates.1 It is derived by the oxidation of dipyrromethane which consists of two pyrrole units bridged through an alpha carbon to nitrogen. Analogously, N-confused dipyrrin consists of a pyrrole unit connected through a beta carbon to the nitrogen. However, the role of N-confused dipyrrin as a metal complexing agent has eluded chemists probably due to its unconventional orientation of the pyrrole rings. Recent reports have recognized that this change in bonding within the dipyrrin framework can significantly alter its reactivity with metal ions. In contrast to regular dipyrrin, N-confused dipyrrin undergoes oxidative coupling in the presence of metal ions to form macrocyclic products known as expanded norroles.2
A norrole is considered to be a structural isomer of a corrole wherein the bipyrrole unit is established through a C–N bond rather than the conventional C–C bond between the adjacent pyrrole units.3 In parallel to this framework, neo-confused porphyrin4 and its derivatives5 have pyrrole nitrogen connected through the meso carbon in the macrocyclic framework. The presence of a terminal N-confused pyrrole in oligopyrrole has been successfully employed to synthesize a corrole, pentaphyrin and hexaphyrin with a C–N bond in the bipyrrolic unit.6 Herein, we report the cyclodimerization of doubly N-confused dipyrrin into an aza derivative of heptalene,7 rather than the expected isomer of a “norcorrole” 1 (Fig. 1).8
 | ||
| Fig. 1 Chemical structures of norcorrole (1) and nornorrole (2). | ||
Following our efforts towards the synthesis of N-confused dipyrrin's metal complex, we attempted to synthesize the metal complex of a dipyrrin derived from 3,3′-dipyrromethane, 3a.6 Its oxidation into 3b (doubly N-confused dipyrrin) was quantitative and reacted with metal salts (Scheme 1) without further purification. The MALDI-TOF mass spectrum of the reaction mixture upon addition of copper(II) acetate to 3b, displayed an m/z value of 615.0664, two units less than the expected “nornorrole” 2 (Fig. 1). It was identified as a pink colored band from column chromatographic separation and isolated in 4% yield. The nature of the products formed from 3b was dependent on the metal salt employed in the reaction. When 3b reacted with iron(III) acetylcetonoate under similar conditions, two different products could be isolated in 5% and 10% yields respectively. Apart from the pink colored band mentioned above, a greenish-yellow band was also isolated from column chromatographic purification. The high-resolution mass spectrum of this band and the pink band displayed the same m/z value, suggesting them as structural isomers of an unexpected cyclodimer.
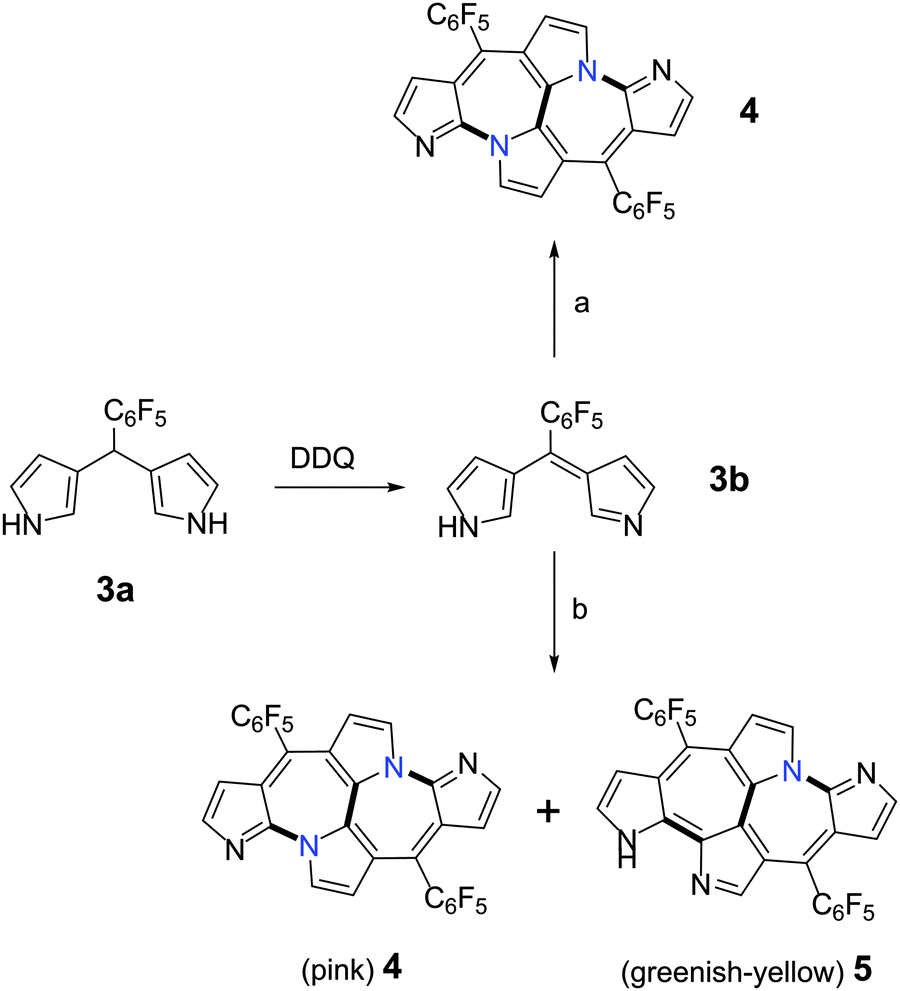 | ||
| Scheme 1 Syntheses of aza-heptalenes, 4 and 5. (a) Cu(OAc)2; (b) Fe(acac)3. (OAc = acetate; acac = acetylacetonoate). | ||
The individual pink and greenish-yellow fractions were allowed to crystallize in dichloromethane by vapor diffusion of n-hexanes. Molecular structures of these compounds obtained from single crystal X-ray diffraction analysis revealed an unusual bicyclic C-fused rings corresponding to the pink and greenish-yellow solutions respectively (Fig. 2).9 The actual structure is an apparent hybrid of a fused bicyclic system, embedded in a macrocyclic framework similar to that of “nornorrole” 2. Perhaps, this may have occurred by the oxidative coupling between the diagonally opposite pyrrole rings in the contracted macrocyclic framework of 2. In a structural similarity to expanded norroles, 4 also exhibited head-to-tail connectivity of two doubly N-confused dipyrrin units (Fig. 2a) leading to the formation of two Cα–N linked bipyrrole units. Both the N-linked pyrrole rings (A and B) have their α-carbon atoms positioned at a distance of 1.413(3) Å. This proximity is sufficient enough for oxidative coupling induced Cα–Cα bond leading to the formation of a third bipyrrolic unit. The co-planarity of the fused seven membered rings suggested the delocalization of the lone pair of electrons on N-linked pyrrole. The flat geometry also aids weak π stacking as observed in its crystal packing (Fig. 2b). 5 revealed an unsymmetrical but a distinct flat structure due to three different bipyrrolic units. The Cα–N and Cα–Cα linkages between the two doubly N-confused dipyrrin units provide an irregular connectivity as compared to 4. An α-carbon of the N-linked pyrrole, C, present in one dipyrrin unit and β-carbon of the other pyrrole, D, present in the other dipyrrin are at a distance of 1.401(7) Å. Oxidative coupling between these two carbon atoms could have induced a bipyrrole unit through an unconventional Cα–Cβ connectivity which aids the unsymmetrical fusion (Fig. 2c). This bond distance is much shorter than the 1.49 Å reported for a similar bond in C-fused norrole.6 All the pyrrole rings and the heptalene10 moiety are planar enough to suggest the delocalization of N-linked nitrogen's lone pair of electrons with the π-electron cloud of the macrocycle (Fig. 2d).
 | ||
| Fig. 2 Molecular structures of aza-heptalenes obtained from single crystal X-ray diffraction analysis. Top views of 4 (a), 5 (c) and side views of 4 (b), 5 (d). In side views, the meso-pentafluorophenyl and hydrogen atoms were omitted for clarity. The C–N bond linkages in the bipyrrole units are highlighted in black. | ||
However, the single crystal X-ray diffraction data were not precise enough to identify the location of the proton with respect to the three different nitrogens in 5. The tautomeric structures suggest possibilities with the hydrogen on any one of the three nitrogens. Hence, we synthesized the N-methyl derivative from 5 to identify the most stable tautomer. The molecular structure of Me-5 determined from single crystal X-ray diffraction analysis confirmed the location of the methyl group on the nitrogen of the bipyrrolic unit formed by the coupling of the alpha carbon atoms (Fig. 3). The structure of this methyl derivative, Me-5, was similar to that of 5 with the formation of a fused bicyclic seven membered ring. All the pyrrole rings and the seven membered rings are planar enough (Fig. 3b) to suggest the delocalization of the lone pair of electrons of N-linked nitrogen with the global π-electron cloud. Substituted heptalenes are known to adopt a non-planar conformation comparable to chiral helicenes.11 However 4 and 5 represent examples of substituted heptalene, whose planarity is favored by the fusion of two pyrrole rings to the central bicyclic core.
 | ||
| Fig. 3 Molecular structure of Me-5 [(a) top view and (b) side view] obtained from single crystal X-ray diffraction analysis. In side views, the meso-pentafluorophenyl and hydrogen atoms were omitted for clarity. The C–N bond linkages in the bipyrrole units are highlighted in black. | ||
All these structures were found to be consistent with their respective 1H NMR spectrum. 4 displayed a symmetrical spectrum with only four doublets at δ 6.84, 7.79, 7.96 and 9.53 ppm corresponding to equal number of protons. In its 1H–1H 2D COSY spectrum, only two correlations between the doublets at (i) 6.84 and 7.79; (ii) 7.96 and 9.53 ppm were observed. This is attributed to the two different sets of protons on the Cα and Cβ of the four pyrrole rings. In contrast to the 1H NMR spectrum of 4, 5 displayed an unsymmetrical spectrum. A total of eight signals in the region δ 6.00 to 13.50 ppm were observed at room temperature. Six doublets at δ 6.28, 6.73, 7.19, 7.50, 7.82 and 9.28 ppm in addition to a sharp singlet at 8.71 ppm and a broad singlet at 13.46 ppm corresponded to equal number of protons. Upon addition of D2O, the intensity of the signal at 13.46 ppm reduced drastically suggesting its identity as the lone NH in the molecule. In its 1H–1H 2D COSY spectrum three correlations were observed between the six doublets. The lone singlet at 8.71 ppm could be identified as the hydrogen on the Cα of the “D” pyrrole ring, while the doublets can be attributed to each individual proton of the macrocycle. DEPT-90 measurement displayed seven 13C signals for the CH at 108.72, 112.25, 112.80, 126.97, 130.80, 140.26, and 149.92 ppm (see the ESI†) that matched the structure of 5. The HMBC spectrum was also found to be in agreement with the above analysis (ESI†). The 1H NMR spectrum of Me-5 exhibited a total of eight signals. A singlet corresponding to three protons at 4.75 ppm was attributed to the protons of the methyl group while the other seven signals resonated with similar chemical shift values as observed for 5. The 1H–1H 2D COSY spectrum also displayed similar correlations as observed for 5 (see the ESI†). The observed downfield chemical shift values signify a diatropic ring current effect for both 4 and 5. However, the expected nornorrole should be a 20π antiaromatic system with considerable upfield chemical shift values. The diatropic ring current effect suggests 4 and 5 as derivatives of substituted heptalene, which are found to be aromatic in nature. This understanding was further supported by electronic absorption spectroscopy and quantum mechanical calculations.
The bond lengths in the optimized structures of 4 and 5 were in agreement with the experimentally determined molecular structures. Their electronic absorption spectrum was found to be almost similar to each other, but significantly different from norcorrole.8 They exhibit multiple absorptions and are relatively blue shifted compared to norcorrole (see the ESI†). The pink colored solution of 4 in dichloromethane exhibited absorptions at 262 nm (ε = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800), 280 (17800), 361 (10000) and 514 (6700). The greenish-yellow colored solution of 5 absorbed at 268 (32600), 282 (32500), 346 (33600) and 451 nm (8800). Absorptions with similar energy were observed for the methyl derivative of 5 in dichloromethane. These absorption values are more comparable to high energy absorptions of heptalene like systems. Ideally porphyrinoids exhibit absorptions in the visible part of the electromagnetic spectrum followed by weak and low energy absorptions. TD-DFT calculations were employed to simulate the electronic absorption spectrum for 4 and 5. At first it was calculated for the whole molecule and later by trimming to the heptalene core of the molecule (ESI†). It was evident that the high energy absorptions were characteristic of heptalene, rather than that of the expected 20π nornorrole structure, 2. We employed Gaussian 0312 to estimate the nucleus independent chemical shift (NICS)13 values for 4 and 5 for the unusual fusion of the seven membered rings at the center of a macrocycle. The estimated NICS(0) for both the rings of the fused bicyclic system in 4 was found to have a value of −0.5 ppm at their respective centers. However, the value differed for both the rings in 5 and was found to be −5 ppm for the heterocyclic seven membered ring and +2 ppm for the carbon only ring. These values are suggestive of a relatively effective π delocalization in the bicyclic system of 5 compared to weak ring currents of 4. Cyclic voltammetry studies displayed redox features uncharacteristic of a porphyrin-like macrocycle. Three reductions were observed at −0.78, −1.04 and −1.24 V for 4 while oxidation could not be identified in its cyclic voltammogram. Similar such reductions at −0.88, −1.1 and −1.46 V along with a single irreversible oxidation at 1.25 V were observed for 5. These studies only suggest the inability of these systems to undergo oxidation and they do not ligate metal ions untypical of porphyrinods.
800), 280 (17800), 361 (10000) and 514 (6700). The greenish-yellow colored solution of 5 absorbed at 268 (32600), 282 (32500), 346 (33600) and 451 nm (8800). Absorptions with similar energy were observed for the methyl derivative of 5 in dichloromethane. These absorption values are more comparable to high energy absorptions of heptalene like systems. Ideally porphyrinoids exhibit absorptions in the visible part of the electromagnetic spectrum followed by weak and low energy absorptions. TD-DFT calculations were employed to simulate the electronic absorption spectrum for 4 and 5. At first it was calculated for the whole molecule and later by trimming to the heptalene core of the molecule (ESI†). It was evident that the high energy absorptions were characteristic of heptalene, rather than that of the expected 20π nornorrole structure, 2. We employed Gaussian 0312 to estimate the nucleus independent chemical shift (NICS)13 values for 4 and 5 for the unusual fusion of the seven membered rings at the center of a macrocycle. The estimated NICS(0) for both the rings of the fused bicyclic system in 4 was found to have a value of −0.5 ppm at their respective centers. However, the value differed for both the rings in 5 and was found to be −5 ppm for the heterocyclic seven membered ring and +2 ppm for the carbon only ring. These values are suggestive of a relatively effective π delocalization in the bicyclic system of 5 compared to weak ring currents of 4. Cyclic voltammetry studies displayed redox features uncharacteristic of a porphyrin-like macrocycle. Three reductions were observed at −0.78, −1.04 and −1.24 V for 4 while oxidation could not be identified in its cyclic voltammogram. Similar such reductions at −0.88, −1.1 and −1.46 V along with a single irreversible oxidation at 1.25 V were observed for 5. These studies only suggest the inability of these systems to undergo oxidation and they do not ligate metal ions untypical of porphyrinods.
In summary, doubly N-confused dipyrrin undergoes intermolecular dimerization with metal salts to form substituted heptalene isomers. Dipyrrin and its derivatives are useful building blocks to construct macrocyclic structures through intermolecular bipyrrole units. Yet 4 and 5 exhibit a molecular structure that stems from the probable macrocyclic intermediate, such as 2, to form an intramolecular bipyrrolic unit at its center to fashion a heptalene moiety. The synthesis is relatively simple in comparison to the multistep synthesis reported for such fused cyclic systems. Even though they represent structural features of N-confused corrole, norrole and norcorrole, spectroscopic characterization and computational studies support 4 and 5 as nonbenzenoid aromatic molecules.
Authors thank SERB-DST, New Delhi, India, and IISER, Pune, for the financial support. BKR thanks the UGC, New Delhi, India, for a senior research fellowship. We thank Prof. M. Ravikanth, IIT-B, Mumbai, India, for the cyclic voltammetric studies.
Notes and references
- T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS PubMed and references cited therein.
- S. C. Gadekar, B. K. Reddy and V. G. Anand, Angew. Chem., Int. Ed., 2013, 52, 7164–7167 CrossRef CAS PubMed.
- K. Fujino, Y. Hirata, Y. Kawabe, T. Morimoto, A. Srinivasan, M. Toganoh, Y. Miseki, A. Kudo and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 6855–6859 CrossRef CAS PubMed.
- T. D. Lash, A. D. Lammer and G. M. Ferrence, Angew. Chem., Int. Ed., 2011, 50, 9718–9721 CrossRef CAS PubMed.
- R. Li, A. D. Lammer, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2014, 79, 4078–4093 CrossRef CAS PubMed.
- (a) Y. Xie, P. Wei, X. Li, T. Hong, K. Zhang and H. Furuta, J. Am. Chem. Soc., 2013, 135, 19119–19122 CrossRef CAS PubMed; M. Toganoh, Y. Kawabe and H. Furuta, J. Org. Chem., 2011, 76, 7618–7622 Search PubMed; (b) P. Wei, K. Zhang, X. Li, D. Meng, H. Agren, Z. Ou, S. Ng, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2014, 53, 14069–14073 CrossRef CAS PubMed.
- H. J. D. Dauben and D. J. Bertelli, J. Am. Chem. Soc., 1961, 83, 4659–4660 CrossRef.
- T. Ito, Y. Hayashi, S. Shimizu, J. Y. Shin, N. Kobayasi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS PubMed.
- CCDC 1024460 (4), 1024461 (5) and 1024462 (Me-5).
- (a) C. Jutz and E. Schweiger, Angew. Chem., Int. Ed., 1971, 10, 808–809 CrossRef CAS PubMed; (b) A. G. Anderson Jr., A. A. MacDonald, A. F. Montana and W. Shive, J. Am. Chem. Soc., 1968, 90, 2993–2994 CrossRef.
- X. Jin, A. Linden and H. J. Hansen, Helv. Chim. Acta, 2010, 93, 729 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 03 (Revision C.02), Gaussian, Inc., Wallingfort, CT, 2004 Search PubMed. For full reference, see the ESI†.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and coordinates for the DFT optimized structures. CCDC 1024460–1024462. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01367d |
| This journal is © The Royal Society of Chemistry 2015 |