
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA–ligand interactions gained and lost: light-induced ligand redistribution in a supramolecular cascade†
Daria V.
Berdnikova
*ab,
Tseimur M.
Aliyeu
b,
Thomas
Paululat
a,
Yuri V.
Fedorov
b,
Olga A.
Fedorova
b and
Heiko
Ihmels
a
aDepartment Chemie–Biologie, Organische Chemie II, Universität Siegen, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: berdnikova@chemie-bio.uni-siegen.de; daria@ineos.ac.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilova str. 28, 119991 Moscow, Russia
First published on 16th February 2015
Abstract
A supramolecular five-component cascade is presented that enables light-controlled transport of an in situ modified ligand between three host systems based on the different complexation preferences of cyclodextrin, cucurbituril, and double-stranded DNA. The results point out novel approaches for the control of drug–DNA interactions in DNA-targeting therapy.
Controlled delivery and release of drugs in a target organ or tissue are key challenges of modern medicine and pharmacology.1 Specifically, DNA-targeting chemotherapy requires highly efficient and selective modes of operation to minimize damage to the healthy cells. In this regard, encapsulation of drugs within macrocyclic “containers” is one of the most successful approaches allowing realization of targeted delivery and release with minimal side effects.2 Cyclodextrins (CD) and cucurbiturils (CB[n]) represent host molecules of major importance due to their low toxicity, chemical stability, and large association constants of the host–guest complexes.3,4
On-demand drug release from the host carrier can be realized by activation of the encapsulated drug molecules with various external triggers, e.g. changes in temperature, pH and oxidation states, variations in electric or magnetic fields, displacement by a competing ligand, or irradiation. In this respect, light is a non-invasive stimulus that enables local and temporal control without interfering with physiological media. Several CD-containing systems for photocontrolled drug release have been reported that operate by light-induced isomerization of azobenzene inside the CD cavity.5,6 However, in these cases the photoreaction provides just a mechanical output resulting in the release of loaded drug molecules from carriers and does not lead to the formation of drug species from the complexed precursors. Surprisingly, there are only a very few examples of “non-azobenzene” systems with light-triggered guest release directly from the CD or CB[n] cavity; and these ones have not been considered for biological purposes.7
Although interactions of ligand–CD8 and ligand–CB[n]9 complexes with DNA have been reported already, in these cases the ligand distribution is governed by the chemical equilibria between interacting components, i.e. no external stimuli were applied to trigger a DNA-binding event in these systems. Moreover, established CD- or CB[n]-comprising supramolecular DNA-binding systems included a maximum of three interactive components, presumably because of the potential ambiguity of the interaction pattern.8,9
Herein, we present for the first time a supramolecular reaction cascade consisting of five components that enables the light-controlled in situ generation and redistribution of a DNA-binding ligand between different host systems. The key process that provides a temporal and spatial control of the formation of a DNA-binder is the photocyclization reaction of styrylpyridine derivative 1, followed by aerobic oxidation to give benzo[c]quinolizinium 2 (Scheme 1). In analogy to the properties of a structurally resembling styrylbenzothiazole10a we proposed that pyridine derivative 1 does not interact with DNA, whereas the photocyclization product 2 is an efficient DNA-intercalator. Furthermore, compound 2 resembles the structure of berberine-type alkaloids11a or phenanthridinium derivatives11b that exhibit remarkable biological activities. The photocyclization may be performed directly in the presence of the nucleic acid resulting in the photoinduced interaction with DNA. In this context, it should be noted that examples of compounds whose DNA-binding properties can be controlled by light are rather rare.10 The delivery, photoinduced transformation and redistribution of the ligand shall be performed based on a delicate balance between different host–guest interactions and binding affinities of the substrates 1 and 2 with (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD), calf thymus DNA (ct DNA) and cucurbit[7]uril (CB[7]) (Scheme 1). Therefore the structures of substrate 1 and resulting photoproduct 2 were chosen such that the relevant associative interactions with the HP-β-CD and CB[7] host systems are orthogonal. In the first step of the reaction sequence, the encapsulation of the ligand 1 in the HP-β-CD cavity provides enhanced solubility of the hydrophobic precursor 1 (step 1). The photocyclization of 1 inside HP-β-CD leads to the irreversible formation of the charged photoproduct 2 that is subsequently released from the hydrophobic HP-β-CD interior (step 2). If DNA is present in the solution, ligand 2 intercalates into the nucleic acid (step 3). Finally, the amount of the DNA-bound intercalator 2 may be manipulated by a concurrent redistribution equilibrium with CB[7] (step 4); i.e., ligand 2 can be extracted from DNA by association with increasing amounts of CB[7].
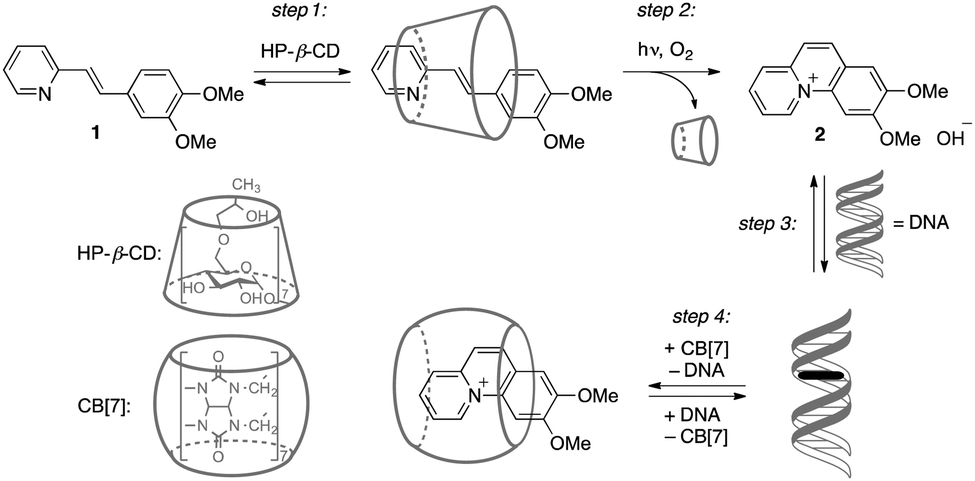 | ||
| Scheme 1 Association and redistribution equilibria of ligands 1 and 2 in the presence of hosts. | ||
Owing to the hydrophobic effect of the cyclodextrin interior, styrylpyridine derivative 1 forms an inclusion complex with HP-β-CD. Although the addition of HP-β-CD to a solution of ligand 1 led to only very small changes in the ligand absorption (Fig. S1, ESI†), fluorimetric titrations demonstrated an increase of the emission intensity of 1 by a factor of 2.8 along with a pronounced blue shift of the emission maximum (Δλ = 28 nm, Fig. S2, ESI†). In addition, a distinct induced circular dichroism (ICD) signal appeared, that resembles the long-wavelength absorption of 1, indicating inclusion of the achiral ligand inside the chiral cyclodextrin host (Fig. S3, ESI†).12 The analysis of the fluorimetric and polarimetric titration data revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the complex between 1 and HP-β-CD with the stability constant Kb = 6.3 × 102 M−1. Complementary evidence for the complexation of 1 with HP-β-CD was provided by 2D NMR spectroscopy. Specifically, clear ROE cross-peaks were observed between the signals of the ligand protons and the internal protons of the HP-β-CD cavity (Fig. S4, ESI†). At the same time, styrylpyridine derivative 1 shows just very weak interaction with ct DNA as demonstrated by photometric and fluorimetric titrations as well as by circular dichroism spectroscopy data (Fig. S5–S7, ESI†). Addition of CB[7] to a solution of 1 resulted in the appearance of a weak red shifted shoulder at around 400 nm in the absorption spectrum that was assigned to the protonated ligand inside the CB[7] cavity (Fig. S8, ESI†).13 Notably, the addition of HP-β-CD to a mixture of 1 and CB[7] suppresses the protonation of 1 completely (Fig. S9, ESI†), thus preventing complexation between ligand 1 and CB[7]. Overall, among three studied supramolecular interactions of derivative 1 only the formation of the inclusion complex with cyclodextrin (step 1 in Scheme 1) was found to be effective.
:1 stoichiometry of the complex between 1 and HP-β-CD with the stability constant Kb = 6.3 × 102 M−1. Complementary evidence for the complexation of 1 with HP-β-CD was provided by 2D NMR spectroscopy. Specifically, clear ROE cross-peaks were observed between the signals of the ligand protons and the internal protons of the HP-β-CD cavity (Fig. S4, ESI†). At the same time, styrylpyridine derivative 1 shows just very weak interaction with ct DNA as demonstrated by photometric and fluorimetric titrations as well as by circular dichroism spectroscopy data (Fig. S5–S7, ESI†). Addition of CB[7] to a solution of 1 resulted in the appearance of a weak red shifted shoulder at around 400 nm in the absorption spectrum that was assigned to the protonated ligand inside the CB[7] cavity (Fig. S8, ESI†).13 Notably, the addition of HP-β-CD to a mixture of 1 and CB[7] suppresses the protonation of 1 completely (Fig. S9, ESI†), thus preventing complexation between ligand 1 and CB[7]. Overall, among three studied supramolecular interactions of derivative 1 only the formation of the inclusion complex with cyclodextrin (step 1 in Scheme 1) was found to be effective.
Other than the styrylpyridine 1, the benzo[c]quinolizinium derivative 2 does not bind to HP-β-CD. Hence, addition of HP-β-CD (up to 1000-fold excess) to 2 did not cause any development of an ICD signal in the circular dichroism spectrum (Fig. S10, ESI†). Such as resembling benzoquinolizinium derivatives14 compound 2 exhibits characteristic properties of a DNA-binding ligand (step 3 in Scheme 1). Thus, the photometric titration of 2 with ct DNA resulted in a pronounced redshift (Δλ = 14 nm) and a strong hypochromic effect of the ligand absorption band (Fig. S11, ESI†). The photometric data were analyzed with a Scatchard plot according to the neighbor exclusion model15 to give a binding constant Kb = 1.2 × 105 M−1. Moreover, the association of 2 with ct DNA was confirmed by the development of a characteristic ICD signal in the range of the ligand absorption upon complex formation (Fig. S12, ESI†).12,16 Complementary fluorimetric titration of ct DNA to 2 revealed an efficient quenching of the fluorescence of 2 in the presence of ct DNA (Fig. S13, ESI†). Additional DNA thermal denaturation experiments showed that ligand 2 stabilizes the DNA duplex towards dissociation into single strands, as indicated by the increase of the DNA melting temperature (ΔTm = 6.3 °C at ligand–DNA ratio LDR = 0.5) (Fig. S14, ESI†). The binding mode of 2 with DNA was determined with viscometric titrations that were analysed as a plot of the cubic root of the relative viscosity, (η/η0)1/3, of the solution versus the LDR (Fig. S15, ESI†). Noteworthy, addition of ligand 2 induced an even stronger increase of the viscosity than ethidium bromide that was employed as reference intercalator, which indicates the pronounced stiffening of the DNA strand upon binding of 2. These results show that ligand 2 intercalates into ct DNA, because, other than groove binders, intercalators increase the viscosity of DNA solutions.17 The interaction of 2 with DNA was additionally studied by 1H NMR spectroscopy using the self-complementary DNA oligonucleotide 5′-CGCGAATTCGCG-3′ (Dickerson dodecamer), and the significant upfield shifts of the DNA imino protons supported the intercalative binding mode of compound 2 (Fig. S21c and d, ESI†).18
On the other hand, the quinolizinium derivative 2 can bind to cucurbit[7]uril due to ion–dipole and hydrophobic interactions. Addition of CB[7] to 2 caused a small redshift (Δλ = 4 nm) of the absorption maximum, along with a strong hypochromic effect (Fig. S16, ESI†). The corresponding binding isotherm was fitted to a 1:1 stoichiometry with a binding constant of the 2–CB[7] complex of Kb = 3.2 × 104 M−1. Strong dynamic broadening and significant upfield shifts of all the aromatic proton resonances in the 1H NMR spectrum indicate complete inclusion of molecule 2 in the CB[7] cavity (Fig. S21a and b, ESI†).19 Overall, photoproduct 2 binds efficiently to both ct DNA and CB[7] (steps 3 and 4, Scheme 1). The stability constant of the 2–DNA complex is about one order of magnitude larger than that of the complex with CB[7]. Therefore, the redistribution of 2 between ct DNA and CB[7] may be directed by changing the relative concentrations of the hosts.
For a complete assessment of the interactive supramolecular interplays, it is also necessary to estimate the influence of HP-β-CD and CB[7] on the stability and structure of the double-stranded DNA. Analysis of mixtures of DNA and HP-β-CD host systems with circular dichroism spectroscopy did not show any changes in the shape and intensity of ct DNA signal even with a 1000-fold excess of HP-β-CD (Fig. S17, ESI†). Moreover, thermal DNA denaturation studies revealed just a nominal stabilizing effect of HP-β-CD on the DNA duplex as shown by a small increase of the DNA melting temperature in the presence of HP-β-CD (ΔTm = 1.9 °C at cHP-β-CD/cDNA = 1500, Fig. S18, ESI†). At the same time, addition of CB[7] led to a small decrease of the DNA melting temperature (ΔTm = −1.1 °C at cCB[7]/cDNA = 2) that may be a sign of partial duplex destabilization or single-strand stabilization (Fig. S19, ESI†). However, further addition of CB[7] resulted in the slow increase of the DNA melting temperature (ΔTm = 0.2 °C at cCB[7]/cDNA = 6). Nevertheless, examination by 1H NMR spectroscopy demonstrated that at equimolar CB[7] concentration the DNA duplex maintains its structural features (Fig. S21c and e, ESI†). Interestingly, DNA melting experiments performed for the ternary mixtures HP-β-CD–DNA–CB[7] showed a slight increase of the DNA melting temperature irrespective of the amount of CB[7] (Fig. S20, ESI†). Overall, at the concentration range used in this study, the presence of HP-β-CD and CB[7] does not significantly affect the stability and structure of the double-stranded DNA.
The photocyclization of styrylpyridine derivative 1 to benzo-quinolizinium 2 in phosphate buffer solution (c1 = 20 μM, pH = 7.0) takes place within 30 min upon irradiation with λ > 250 nm (Fig. S22, ESI†). Likewise, the irradiation of the 1–HP-β-CD complex in aqueous medium led to the rapid initial formation of the cis-isomer of 1, as indicated by the change of the absorption and CD bands (Fig. S23, ESI†). The subsequent photoelectrocyclization-oxidation sequence yields the charged product 2 that cannot be accommodated in the hydrophobic environment of HP-β-CD and, therefore, is ejected from the host cavity.20 Importantly, the phototransformation of 1 occurs in the cyclodextrin interior that is indicated by maintenance of an ICD signal until the full conversion of 1 to 2 is reached. The presence of the cyclodextrin does just slow down the photoreaction by a factor of ca. 2. Irradiation of compound 1 in the presence of ct DNA led to characteristic changes in the absorption and CD spectra that clearly indicated the intercalation of the in situ formed photoproduct 2 and ct DNA (Fig. S24, ESI†). Interestingly, the presence of DNA has no influence on the reaction time. When the irradiation of 1 was performed in the presence of both ct DNA and HP-β-CD (Fig. 1A and B), the positive ICD signal in the range of 310–360 nm that corresponds to the 1–HP-β-CD complex vanished, whereas the positive ICD band of the DNA-intercalated ligand 2 appeared at around 400 nm. Under these conditions the reaction time was not affected.
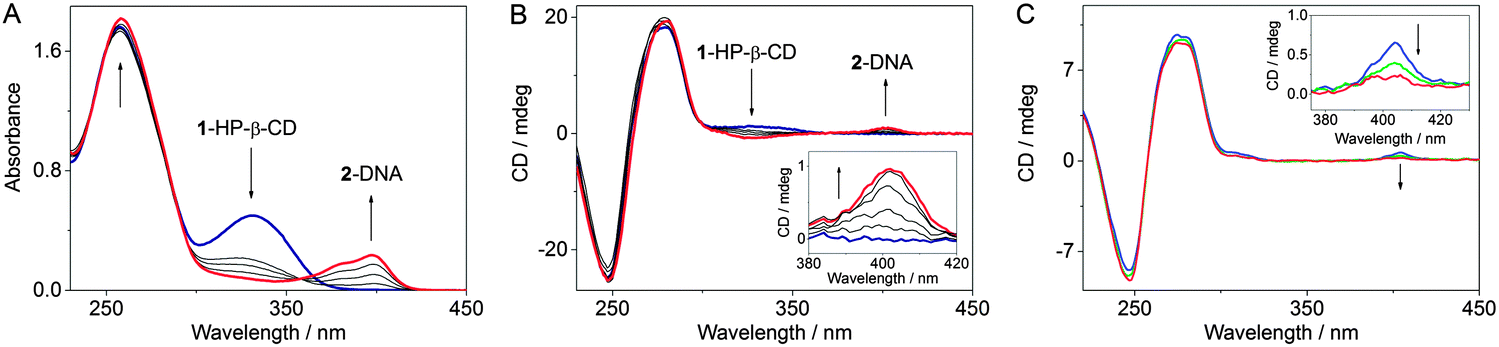 | ||
| Fig. 1 Photocyclization of 1 encapsulated in HP-β-CD in the presence of ct DNA monitored by (A) absorption and (B) CD spectroscopy (c1 = 20 μM, cHP-β-CD = 10 mM, cDNA = 0.1 mM, full light of a high pressure Hg lamp, spectra were recorded every 5 min, timescale 0–30 min). (C) Circular dichroism spectra of 2–ct DNA mixtures (c2 = 15 μM, cDNA = 50 μM) in the presence of 500 eq. of HP-β-CD (cHP-β-CD = 7.5 mM): blue: without CB[7]; green: with addition of 5 eq. of CB[7] (cCB[7] = 75 μM); red: with addition of 10 eq. of CB[7] (cCB[7] = 150 μM). Inset: disappearance of the ICD signal of the 2–DNA complex due to extraction of ligand 2 from the DNA helix by CB[7]. All spectra were recorded in phosphate buffer (pH 7.0) at 20 °C. | ||
In order to study the propensity of CB[7] to extract the ligand 2 from DNA, competition experiments were conducted with CB[7] and DNA. Spectrophotometric analysis indicated that the addition of a 40-fold excess of CB[7] to a solution of ct DNA with intercalated 2 (cDNA/c2 = 8) was accompanied by the development of the characteristic absorption bands of the inclusion complex 2–CB[7], which denotes the extraction of 2 from the DNA host by CB[7] (Fig. S26, ESI†). Notably, the extraction ratio did not change in the presence of 500 eq. of HP-β-CD in the initial mixture. These results were confirmed by disappearance of the ICD signal of the 2–DNA complex (c2/cDNA = 1.5/5) upon addition of 10 molar eq. of CB[7] (Fig. 1C and Fig. S28, ESI†). Unfortunately, in the presence of DNA, a large excess of CB[7] causes precipitation of the 2–CB[7] complex or/and CB[7]. The precipitation was even more pronounced when HP-β-CD (500 eq.) was added to the initial mixture. Complementary experiments showed that the redistribution equilibrium may also be shifted to the formation of the complex between 2 and DNA. Thus, upon addition of ct DNA to a 2–CB[7] mixture (cCB[7]/c2 = 10) the ligand 2 was extracted by DNA at a LDR = 30 (Fig. S27, ESI†). The presence of 500 eq. of HP-β-CD in the initial mixture had no influence on the concurrent binding process and did not affect the extraction ratio as was clearly shown by absorption spectroscopy.
To demonstrate that the complete cascade can be established even in the four-component mixture, we performed the photocyclization in a mixture comprising 1, HP-β-CD, ct DNA, and CB[7] (c1/cHP-β-CD/cDNA/cCB[7] = 1/500/5/10) (Fig. S25, ESI†). In this system, the absorption maximum of the in situ formed 2 adopts an intermediary position (λmax = 394 nm) between the absorption maxima of the 2–CB[7] (λmax = 389 nm) and 2–DNA (λmax = 398 nm) complexes indicating redistribution of the ligand between DNA and CB[7] hosts. Accordingly, the intensity of the ICD signal of the 2–DNA complex in the quaternary system is reduced in comparison with the system without CB[7] under equal conditions, because the photoproduct 2 is partially bound to achiral CB[7].
In summary, we have demonstrated a proof-of-principle for a light-operated supramolecular cascade representing a novel integral approach toward controlled ligand–DNA interactions. The cascade starts with the photoinduced in situ generation of the intercalating molecule from the encapsulated precursor, continues by association of the intercalator with the nucleic acid and finishes with removal of the bound ligand from the DNA binding site. In spite of the simultaneous presence of several host molecules, HP-β-CD, CB[7] and DNA, each step of the cascade is unaffected by the presence of non-involved components. Importantly, the realization of the phototransformation of the precursor 1 into DNA-intercalator 2 inside the cyclodextrin cavity substantially increases biocompatibility of the method. Considering the recently discovered ability of CB[n]s and their complexes to cross the cell membrane21 and to penetrate into the cell nuclei,22 this approach represents a promising method for drug deactivation or/and overdose treatment.
Generous support by the Deutsche Forschungsgemeinschaft and RFBR 14-03-32038 is gratefully acknowledged.
Notes and references
- (a) A. S. Hoffman, J. Controlled Release, 2008, 132, 153 CrossRef CAS PubMed; (b) Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198 CrossRef CAS PubMed; (c) E. R. Balmayor, H. S. Azevedo and R. L. Reis, Pharm. Res., 2011, 28, 1241 CrossRef CAS PubMed.
- C. P. Leamon and P. S. Low, in Drug Delivery: Principles and Applications, ed. B. Wang, T. J. Siahaan and R. A. Soltero, Wiley-Interscience, New York, 2009, ch. 9 Search PubMed.
- K. Uekama, F. Hirayama and H. Arima, in Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications, ed. H. Dodziuk, WILEY-VCH, Weinheim, 2006, ch. 14, pp. 381–422 Search PubMed.
- A. I. Day and J. G. Collins, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. Gale and J. Steed, WILEY-VCH, Weinheim, 2012 Search PubMed.
- (a) D. P. Ferris, Y.-L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686 CrossRef CAS PubMed; (b) R. Liu, Y. Zhang and P. Feng, J. Am. Chem. Soc., 2009, 131, 15128 CrossRef CAS PubMed; (c) D. Tarn, D. P. Ferris, J. C. Barnes, M. W. Ambrogio, J. F. Stoddart and J. I. Zink, Nanoscale, 2014, 6, 3335 RSC; (d) S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Angew. Chem., Int. Ed., 2011, 50, 9747 CrossRef CAS PubMed; (e) J. Zheng, Y. Nie, S. Yang, Y. Xiao, J. Li, Y. Li and R. Yang, Anal. Chem., 2014, 86, 10208 CrossRef CAS PubMed.
- (a) Y. Wang, M. Zhang, C. Moers, S. Chen, H. Xu, Z. Wang, X. Zhang and Z. Li, Polymer, 2009, 50, 4821 CrossRef CAS PubMed; (b) W. Xiao, W.-H. Chen, X.-D. Xu, C. Li, J. Zhang, R.-X. Zhuo and X. Z. Zhang, Adv. Mater., 2011, 23, 3526 CrossRef CAS PubMed; (c) K. Peng, I. Tomatsu and A. Kros, Chem. Commun., 2010, 46, 4094 RSC.
- (a) Y. Kim, Y. H. Ko, M. Jung, N. Selvapalam and K. Kim, Photochem. Photobiol. Sci., 2011, 10, 1415 RSC; (b) V. Petrov, S. Stanimirov, I. K. Petrov, A. Fernandes, V. de Freitas and F. Pina, J. Phys. Chem. A, 2013, 117, 10692 CAS.
- (a) S. Chandrasekaran, Y. Sameena and I. V. Enoch, J. Mol. Recognit., 2014, 27, 640 CrossRef CAS PubMed , and other papers of Enoch's group; (b) H. Y. V. Ching, D. P. Buck, M. Bhadbhade, J. G. Collins and L. M. Rendina, Chem. Commun., 2012, 48, 880 RSC; (c) Y. Shi and J. C. Dabrowiak, Inorg. Chim. Acta, 2012, 393, 337 CrossRef CAS PubMed; (d) J.-P. Song, Y.-J. Guo, Q. Zhao, S.-M. Shuang, C. Dong and M. M. F. Choi, Talanta, 2010, 82, 681 CrossRef CAS PubMed; (e) A. M. Krause-Heuer, N. J. Wheate, M. J. Tilby, D. G. Pearson, C. J. Ottley and J. R. Aldrich-Wright, Inorg. Chem., 2008, 47, 6880 CrossRef CAS PubMed; (f) S. Sato, T. Nojima, M. Waki and S. Takenaka, Molecules, 2005, 10, 693 CrossRef CAS; (g) G. Zhang, S. Shuang, C. Dong, D. Liu and M. M. F. Choi, J. Photochem. Photobiol., B, 2004, 74, 127 CrossRef CAS PubMed; (h) M. S. Ibrahim, I. S. Shehatta and A. A. Al-Nayeli, J. Pharm. Biomed. Anal., 2002, 28, 217 CrossRef CAS; (i) G.-C. Zhao, J.-J. Zhu and H.-Y. Chen, Spectrochim. Acta, Part A, 1999, 55, 1109 CrossRef.
- (a) S. Sun, Y. Yuan, Z. Li, S. Zhang, H. Zhang and X. Peng, New J. Chem., 2014, 38, 3600 RSC; (b) F. Li, Y. Xu, H. Li, C. Wang, A. Lu and S. Sun, New J. Chem., 2014, 38, 1396 RSC; (c) H. Isobe, N. Tomita, J. W. Lee, H.-J. Kim, K. Kim and E. Nakamura, Angew. Chem., Int. Ed., 2000, 39, 4257 CrossRef CAS.
- (a) D. Berdnikova, O. Fedorova, E. Gulakova and H. Ihmels, Chem. Commun., 2012, 48, 4603 RSC; (b) K. Starčević, G. Karminski-Zamola, I. Piantanida, M. Žinić, L. Šuman and M. Kralj, J. Am. Chem. Soc., 2005, 127, 1074 CrossRef PubMed; (c) J. Andersson, S. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836 CrossRef CAS PubMed; (d) M. L. Di Pietro, F. Puntoriero, F. Tuyéras, P. Ochsenbein, P. P. Lainé and S. Campagna, Chem. Commun., 2010, 46, 5169 RSC; (e) S. V. Paramonov, V. Lokshin, H. Ihmels and O. A. Fedorova, Photochem. Photobiol. Sci., 2011, 10, 1279 RSC; (f) M. I. Sánchez, O. Vázquez, M. E. Vázquez and J. L. Mascareñas, Chem. Commun., 2011, 47, 11107 RSC; (g) H. Ihmels, J. Mattay, F. May and L. Thomas, Org. Biomol. Chem., 2013, 11, 5184 RSC.
- (a) Z.-J. Huang, Y. Zeng, P. Lan, P.-H. Sun and W.-M. Chen, Mini-Rev. Med. Chem., 2011, 11, 1122 CrossRef CAS; (b) L.-M. Tumir, M. Radić Stojković and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930 CrossRef PubMed.
- S. Allenmark, Chirality, 2003, 15, 40 CrossRef PubMed.
- N. Barooah, J. Mohanty, H. Pal and A. C. Bhasikuttan, Proc. Natl. Acad. Sci., India, Sect. A, 2014, 84, 1 CrossRef CAS.
- H. Ihmels, K. Faulhaber, D. Vedaldi, F. Dall'Acqua and G. Viola, Photochem. Photobiol., 2005, 81, 1107 CrossRef CAS PubMed.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469 CrossRef CAS.
- B. Norden and T. Kurucsev, J. Mol. Recognit., 1994, 7, 141 CrossRef CAS PubMed.
- D. Suh and J. B. Chaires, Bioorg. Med. Chem., 1995, 3, 723 CrossRef CAS.
- M. R. Keniry and R. H. Shafer, Methods Enzymol., 1995, 261, 575 CAS.
- W. L. Mock and N.-Y. Shih, J. Org. Chem., 1986, 51, 4440 CrossRef CAS.
- Y. V. Fedorov, S. V. Tkachenko, E. Y. Chernikova, I. A. Godovikov, O. A. Fedorova and L. Isaacs, Chem. Commun., 2015, 51, 1349 RSC.
- (a) P. Montes-Navajas, M. González-Béjar, J. C. Scaiano and H. García, Photochem. Photobiol. Sci., 2009, 8, 1743 RSC; (b) V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037 RSC.
- W. Lei, Q. Zhou, G. Jiang, Y. Hou, B. Zhang, X. Cheng and X. Wang, ChemPhysChem, 2011, 12, 2933 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, experimental details, and photochemical data. See DOI: 10.1039/c5cc01025j |
| This journal is © The Royal Society of Chemistry 2015 |