
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene oxide nanocapsules within silanized hydrogels suitable for electrochemical pseudocapacitors†
R.
Kataky
*a,
J. H. L.
Hadden
a,
K. S.
Coleman
a,
C. N. M.
Ntola
a,
M.
Chowdhury
a,
A. R.
Duckworth
a,
B. P.
Dobson
a,
R.
Campos
ab,
S.
Pyner
c and
F.
Shenton
c
aDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK. E-mail: ritu.kataky@durham.ac.uk
bInterdisciplinary Nanoscience Center (iNANO), Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus, Denmark
cSchool of Biological and Biomedical Sciences, University of Durham, South Road, Durham, DH1 3LE, UK
First published on 7th May 2015
Abstract
Soft biocompatible gels comprised of rolled up graphene oxide nanocapsules within the pores of silanized hydrogels may be used as electrochemical pseudocapacitors with physiological glucose or KOH as a reducing agent, affording a material suitable for devices requiring pulses with characteristic time less than a second.
Implantable, portable, electronic devices require rapid charge and discharge rates. Transient electronics, where biodegradable electronics are used in implantable devices, is emerging as a new technology.1–4 Electrochemical supercapacitors are emerging for improving the life cycle and charge discharge rate of hybrid biofuel cell devices.5 It is possible that future electrochemical supercapacitors may replace microbatteries in electrochemical energy storage systems where high power delivery is required in short times. Materials for short term implantable devices should ideally be non-toxic, biocompatible and biodegradable, and be capable of rapid charge discharge rates for monitoring transient biological events.1
Rolled-up graphene oxide (GO) is an emerging material of which there are only a handful of reports. Polyvinyl alcohol (PVA)–GO nanocomposites have been widely reported, particularly as a means of improving the mechanical properties of the composite materials, attributed to the hydrogen bonding of the PVA chains to GO.7–9 Rolled up structures of 2D GO forming carbon nanoscrolls (CNS) has previously been reported by Wang and co-workers10 as an emerging carbon nanomaterial. The nano/microscroll structure was attributed to Ag and Fe3O4 nanoparticle aggregation behaviour mainly attached to the edge of the GO. Feng and co-workers,11 recently reported rolled up GO sheets formed by evaporating GO from dispersions onto aluminium foil placed 4 cm above the aqueous dispersion. The authors attributed the formation of these rolled up structures to strong interactions between GO and the alumina layer on the sheet. The structure could be controlled by adjusting the evaporation time and temperature.
It has been reported that thin films incorporated with nanostructured redox active materials such as MnO2, RuO2 and V2O5 nanostructures allow combination of an electric double layer and a Faradic reaction at the surface of the nanoparticles, leading high specific capacitances upto 1200 F g−1 at a scan rate of 2 mV s−1.12,13 In this work, we demonstrate that rolled up GO structures are very suitable for implantable medical devices and stretchable devices that require pulses in the sub second ranges.14,15 These materials are able to form redox pseudocapacitors with fast reversible surface redox reactions for charge storage.16 The specific capacitances are comparable and higher than to recent ZnO and SnO2 nanowires, carbon fibre hybrid devices reported recently for wearable, stretchable devices (12 mF cm−2).15
Porous and activated carbons, carbon nanotubes and graphene have been used as electrode materials for supercapacitors.9 GO4,5 is the oxygenated counterpart for graphene consisting of oxygen based functional groups (e.g. hydroxyls, diols, ketones and carboxyls) covalently bonded to the basal planes and edges of the carbon atoms. While the precise chemical structure of GO is under discussion, experiments suggest that tertiary alcohols, ethers and epoxides are the dominant functional groups on GO6,8,10 with a carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxygen ratio of approximately 2. GO is electrochemically inert but exhibits reduction peaks at approximately −700 mV (vs. Ag/AgCl),7 the accurate reduction potential being dependent on the method for preparing GO, which greatly influences its redox characteristics due to the variations in oxygen functionality.7 The oxygen functional groups make GO hydrophilic and readily dispersed in water.8 Kim and co-workers17 have shown that GO can exhibit pH dependent amphiphilic behaviour. PVA is a hydrogel which is biocompatible and non-toxic, widely used in a range of applications10,11,15 and is known to achieve molecular dispersion of GO in an aqueous phase.
:oxygen ratio of approximately 2. GO is electrochemically inert but exhibits reduction peaks at approximately −700 mV (vs. Ag/AgCl),7 the accurate reduction potential being dependent on the method for preparing GO, which greatly influences its redox characteristics due to the variations in oxygen functionality.7 The oxygen functional groups make GO hydrophilic and readily dispersed in water.8 Kim and co-workers17 have shown that GO can exhibit pH dependent amphiphilic behaviour. PVA is a hydrogel which is biocompatible and non-toxic, widely used in a range of applications10,11,15 and is known to achieve molecular dispersion of GO in an aqueous phase.
A simple method of preparing GO nanocapsules is used where the GO is allowed to ‘rollup’ within the pores of silanized PVA hydrogels, forming gels with discrete GO capsules dispersed within the matrix. 33 mg of dried graphene oxide was dispersed in 15 mL of distilled water and sonicated for 2 hours. A separate PVA solution was prepared by dissolving 3.25 g of PVA in 100 mL of deionized water at 90 °C and then cooling to 50 °C. To this solution at 50 °C, 15 mL of GO solution was added (PVA–GO gel). A gel without GO was prepared using distilled water (PVA gel), for comparison. This mixture was then sonicated for 30 minutes and allowed to dry on a Teflon petri dish for two days at 60 °C until a stable weight was achieved. The procedure was repeated using different amounts of GO to produce hydrogels of 1:10, 1:7.5 and 1:5 ratios of GO to PVA. The fluorination of the gels was performed by immersing the gels in a 50% (w/w) solution of 1H,1H,2H,2H-perfluorodecyltrichlorosilane (PF–SiCl3) in methyl ethyl ketone (MEK) for 48 hours at room temperature, followed by rinsing with MEK repeatedly to remove any physically adsorbed PF–SiCl3 and drying at 60 °C for 24 hours.18
The hydrogel of 1:10 ratio GO to PVA was characterised using contact angle measurements (Fig. 1), X-ray diffraction (XRD), attenuated total reflectance Fourier transform infra-red spectroscopy (ATR-FTIR), Raman and differential scanning calorimetry (DSC) (ESI,† Fig. S1–S3 and S4c, including experimental details) and thermal analyses (ESI,† Fig. S4a and b) confirm the presence of GO in the silanized gels.
 | ||
| Fig. 1 cryo-SEM images and contact angle measurement of different gels (a) PVA–GO (non silanized), hydrophilic, (b) PVA (Si), hydrophobic and (c) PVA–GO (Si), hydrophobic. | ||
The XRD patterns of the PVA–GO (Si) film showed a single sharp peak at 2θ = 17.2°. This has been shifted compared to PVA (Si) film which showed a single peak at 2θ = 19.5° indicating individual dispersion of GO in the films. The shift relative to the PVA–GO peak, at 2θ = 19.9°, shows an increase in layer spacing on fluorination.6 No peak for GO spacing was seen in the composites, due to the low GO:PVA ratio, shifting the peak to below the scanning range.19 ATR-FTIR spectra revealed the successful silanization of the gels. The broad band, seen in PVA and PVA–GO spectra at 3270/3280 cm−1 respectively (O–H stretching vibrations from water molecules and hydrogen bonded PVA), is lost in the silanized gel spectrum due to the reaction of the hydroxyl groups with the fluoroalkyl silane, forming Si–O groups and a large characteristic peaks at 1200 cm−1 and 1145 cm−1.19 While PVA peaks dominate the PVA–GO spectrum due to the low GO:PVA ratio, shifts in characteristic features to higher wavenumbers, such as the band at 3280 cm−1, show the incorporation and strong hydrogen bonding of the GO to the PVA.6 The Raman spectrum of the background matrix confirmed the silanization of the PVA gel. Raman mapping of the PVA–GO (Si) membrane clearly shows the D and G bands at ∼1330 cm−1 and 1590 cm−1 (peak ratio 1:3), with no 2D band observed.
Thermal stability studies of pure PVA showed an onset of degradation at about 300 °C. For the GO loaded gels, the first step is at 285 °C is due to the elimination of the oxygen containing moieties on the GO. The onset of main-chain pyrolysis for the GO gels occurs at a slightly higher temperature compared to the unloaded PVA, indicating increased thermal stability due to strong hydrogen bond interaction and agrees with the FTIR data. Increasing area under the DSC thermograms from PVA to PVA–GO and PVA–GO (Si) suggest increasing crystallinity.
Morphological studies of the gels were performed by cryo-SEM. Small pieces of the hydrogel, with a thickness of approximately 1 mm, were cut with a scalpel and handled with a pair of tweezers. The samples were then fast frozen by plunging in a liquid nitrogen-cooled liquid propane container. The frozen and brittle gel sample was quickly transferred into the liquid nitrogen compartment, fractured with a scalpel blade and transformed into powder-like fine pieces. The resulting doped gel particles were then floated in liquid nitrogen. An amorphous copper SEM grid was submerged into the liquid nitrogen, capturing small gel particles on the grid. This procedure preserved the gel in a hydrated form by rapid freezing. The samples were imaged using an SEM (Hitachi S5200 High-Resolution Field Emission SEM) cryostage maintained at 190 °C. In order to study the structure of the polymer network, the hydrogel sample was freeze-dried in situ by raising the temperature to 100 °C and then the imaging was carried out at a scanning voltage of 1.5 kV. The cryo-SEM images of the freeze dried, PVA–GO (Si) composites showed evidence of tubular structures of rolled up GO capsules (Fig. 1a). These rolled up structures had diameters of up to 300 nm and were of variable length. Some rolled up structures were branched. Control experiments with non-silanized PVA–GO hydrogels, showed images that are very similar to images of PVA–GO composites reported previously,20 no rolling-up behaviour was observed.
Static contact angle measurements were performed by placing a deionised water droplet on a film of the PVA (Si), and PVA–GO (Si) gels. The water droplet static contact angles (θ) were 92° and 112° respectively (Fig. 1b and c), and were stable for at least 2 minutes. The water droplet contact angle on the non-silanized PVA–GO membrane (Fig. 1a) was very small, at θ ∼ 20°, and unstable (not shown).
High resolution transmission microscope (HRTEM) images were obtained by depositing a thin film of the PVA–GO (Si) on holey carbon grids. Low magnification TEM images and selected area electron diffraction (SAED) images were obtained using a JEOL 2100F FEG TEM with a Gatan SC-1000 Orius CCD camera at 200 kV energy. SAED images on a selected spot was obtained at 80 kV in a JEOL 2010F fitted with a CEOS aberration corrector with the spherical aberration coefficient C3 tuned to +1 μm (Fig. 2).
 | ||
| Fig. 2 (a) TEM image of a ‘rolled up’ graphene oxide wire in the silanized PVA–GO (Si) gels, (b) high resolution TEM (HRTEM) and (c) selected-area electron diffraction pattern images. | ||
SEM (Fig. 1b, ESI,† Fig. S5d), TEM (Fig. 2a), high resolution TEM (HRTEM) (Fig. 2b), and selected-area electron diffraction pattern images (Fig. 2c) confirmed the ‘rolling up’ of GO in the silanized gels. The interlattice spacings of approximately 0.36 nm are indicative of graphene layers. Clear diffraction spots indicative of a hexagonal lattice are clearly seen (an enlarged image is available in ESI,† Fig. S5c). Low magnification TEM images (ESI,† Fig. S5a and b) show GO as a highly electron transparent film.21 Layer formation via the self-assembly of alkyl silanes with hydroxyl groups is also a well-known phenomenon,22 which may influence the structures observed. It is likely that the rolled-up structures seen here are a result of the repulsive forces between the hydrophobised pores of the PVA gels and the hydrophilic moieties of the GO sheets which makes it thermodynamically favourable for the GO sheets to roll up. Favourable π–π interactions caused by the overlap of the GO sheets are an additional driving force for decreasing the free energy which counteracts the increased energy from curvature.18 Additionally, GO is an amphiphile,22–24 its basal plane is a network of hydrophobic polyaromatic benzene rings and the edges tend to be hydrophilic. In contrast, the control PVA (Si), showed featureless SEM images (Fig. 1a).
The applicability of the PVA–GO (Si) gels as biocompatible redox supercapacitors was tested using cyclic voltammetry (not shown here, ESI,† Fig. S6) and galvanostatic charge–discharge studies. Electrochemical studies were performed using glassy carbon (GC) electrodes (BASi, A = 0.032 cm2), which were thoroughly cleaned with alumina powder (BASi, 0.05 mμ). The electrodes were then prepared by drop casting 50 μL of the PVA or PVA–GO hydrogel solution onto the electrode surface and allowed to dry at room temperature for 48 h, followed by fluorination. A potentiostat (Ivium, CompactStat) interfaced with a personal computer was used for the electrochemical measurements. A three-electrode cell containing a Pt wire pseudo-reference electrode, a Pt foil (area, 1 cm2) counter electrode and a GC working electrode with the gel drop-casted was used. The cell was placed in a Faraday cage to eliminate external interference. Measurements of the silanized gels were taken using aqueous solutions of 6.0 M KOH(aq), and 0.5 M glucose in phosphate buffered saline (PBS).16 The reduction of GO using glucose as a reducing agent under basic conditions has been observed by Zhu and co-workers24 and Zhang and co-workers25 respectively.
Charge–discharge measurements (Fig. 3) were taken after the gel casted GC electrode was submerged in the electrolyte solution for 3 hours. The peak current pulse was varied depending on the sample response so the potential did not exceed a 5 V potential window. Responces over long and short time scales were studied. The current was cycled at a frequency of 2.5 Hz in order to mimic the potential demands for a small implantable device.12 The profiles obtained show rapid charging and discharging as well as no loss in efficiency after numerous cycles. The response is also easily tunable by altering the GO loading in the hydrogel. Increasing the GO loading and reducing the GO using KOH increased the specific capacitance of the PVA–GO (Si). The capacitance for the 1:5 GO to PVA ratio hydrogel range from 100–260 mF g−1. In a previous report Bae et al. reported a value of largest area specific capacitance of 12 mF cm−2 for a SnO2 nanowire grown on carbon paper for potential applications in wearable and stretchable fabric based supercapacitors.14 The PVA–GO (Si) composites are therefore useful for implantable or wearable devices.14,25,26 Preliminary studies were also conducted with the 1:5 PVA–GO (Si), hydrogels using citrate as a reducing agent. Initial results indicate citrate as a good, alternative reagent for the reduction of PVA GO (Si) composites (ESI,† Fig. S6).
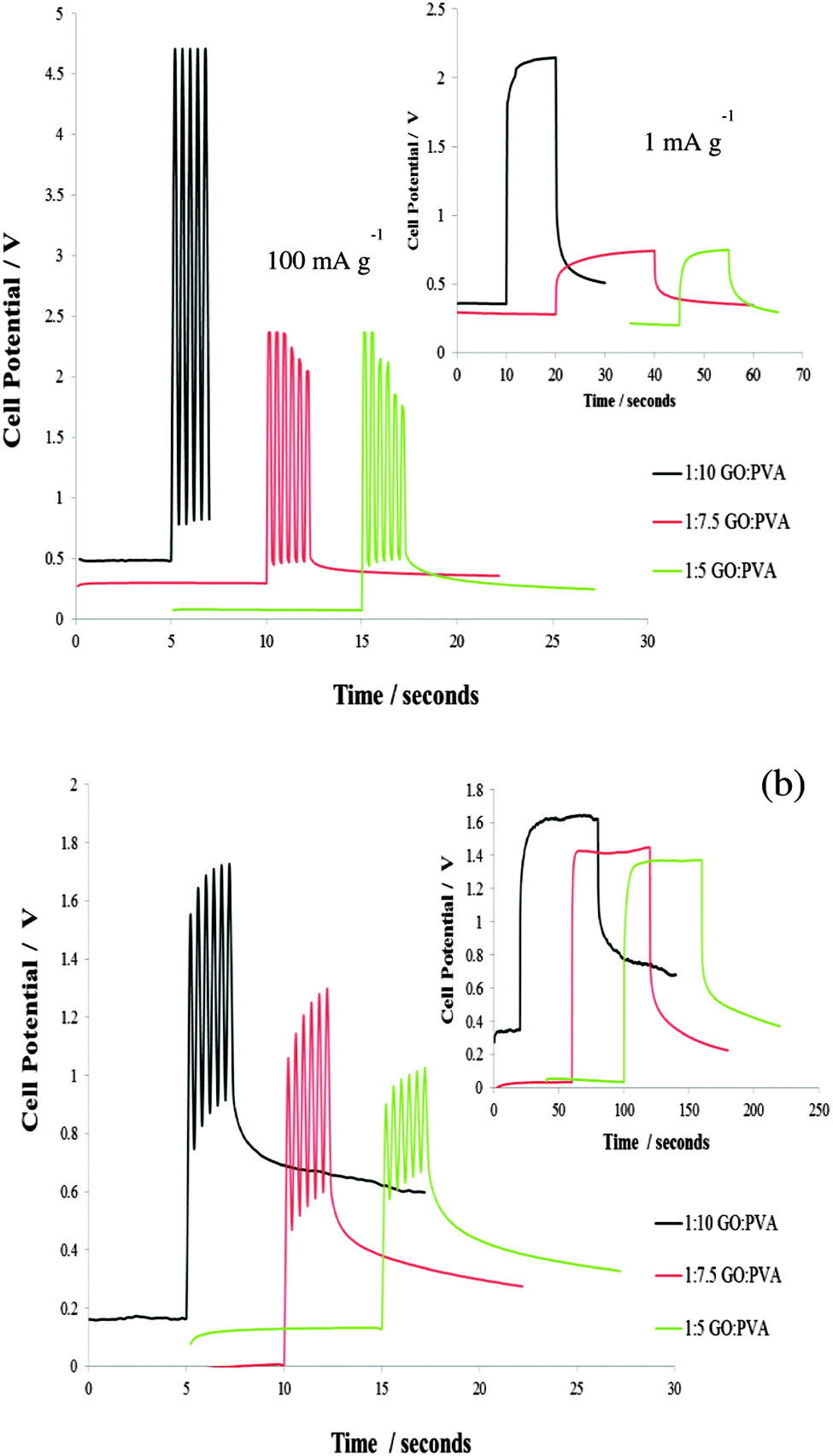 | ||
| Fig. 3 (a) Response in Glucose PBS – 100 mA g−1 0.2 s excitation for fast response and 1 mA g−1 20 s excitation for slow response (b) response in 6.0 M KOH – 20 mA g−1 excitation for fast (0.2 s) and slow (60 s) response. | ||
The biocompatibility of the PVA–GO (Si) was preliminarily assessed by implantation studies (ESI,† Fig. S7, including experimental details). Two pieces of membrane, 25 mm2, were placed under the skin of a male rat and then removed after 7 weeks. The gel remained robust and maintained its integrity, with no adverse effects in the rats. In previous work pegylated GO composites have been reported as water soluble, biocompatible cargos for non-covalently attached, π–π stacked hydrophobic drugs.27 The GO–PVA (Si) composites are not water soluble and remained intact during the 7 week study. This is encouraging for further studies to confirm biocompatibility.
GO entrapped within silanized PVA hydrogels form nanocapsule like structure, with surface redox activity. Characterisation and morphological studies of the composites have confirmed the presence of the GO and its “rolled up” structure in the silanized gels. Electrochemical studies have shown the ability of the gel to rapidly charge and discharge in physiological solutions such as glucose and in KOH, making the material suitable for use in devices for monitoring transient biological events such as electrochemical supercapacitors and stretchable, wearable devices.
Our acknowledgement to B. Mendis (Department of Physics, Durham University) for the TEM microscopy work and EPSRC for funding Rui Campos's studentship (EP/F01189X/1).
Notes and references
- L. Yin, X. Huang, H. Xu, Y. Zhang, J. Lam, J. Cheng and J. A. Rogers, Adv. Mater., 2014, 26, 3879–3884 CrossRef CAS PubMed.
- Y. J. Kim, S. E. Chun, J. Whitacre and C. J. Bettinger, J. Mater. Chem. B, 2013, 1, 3781 RSC.
- H. Jimbo and N. Miki, Sens. Actuators, B, 2008, 134, 219 CrossRef CAS PubMed.
- Y. J. Kim, W. Wu, S. E. Chun, J. F. Whitacre and C. J. Bettinger, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20912 CrossRef CAS PubMed.
- Y. J. Kang, S. J. Chun, S. Suk Lee, B. Y. Kim, J. H. Kim, H. Chung, S. Y. Lee and W. Kim, ACS Nano, 2012, 6(7), 6400–6406 CrossRef CAS PubMed.
- H. Feng, Y. Li and J. Li, RSC Adv., 2012, 2, 6988–6993 RSC.
- J. J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. F. Ma, T. Y. Guo and Y. S. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS PubMed.
- B. Das, K. E. Prasad, U. Ramamurty and C. N. R. Rao, Nanotechnology, 2009, 20, 125705 CrossRef PubMed.
- L. Zhang, Z. Wang, C. Xu, Y. Li, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 10399–10406 RSC.
- S. Wang, D.-P. Yang, G. Huang, P. Huang, G. Shen, S. Guo, Y. Mei and D. Cui, J. Mater. Chem., 2012, 22, 17441–17444 RSC.
- K. Feng, Y. Cao and P. Wu, J. Mater. Chem., 2012, 22, 11455–11457 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS PubMed.
- J. Bae, Y. J. Park, J. C. Yang, H. W. Kim and D. Y. Kim, J. Solid State Electrochem., 2015, 19, 211–219 CrossRef CAS PubMed.
- R. A. Huggins, Solid State Ionics, 2000, 134, 179–195 CrossRef CAS.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and W. Ma, Carbon, 2011, 49, 573–580 CrossRef CAS PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- T. Nishino, M. Meguro and K. Nakamae, Int. J. Adhes. Adhes., 1999, 19, 399–403 CrossRef CAS.
- D. R. Anderson, in Analysis of Silicones, ed. A. L. Smith, Wiley, New York, 1974, ch. 10, vol. 1 Search PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- N. R. Wilson, P. P. Pandey, R. Beanland, R. J. Young, I. A. Kinloch, L. Gong, Z. Liu, K. Suenaga, J. P. Rourke, S. J. York and J. Sloan, ACS Nano, 2009, 9, 2547–2556 CrossRef PubMed.
- S. F. Braga, V. R. Coluci, S. B. Lgoas, R. Giro, D. S. Galvao and H. Baughman, Nano Lett., 2004, 4, 881–884 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowsk, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang, B.-Y. Kim and S. Dong, ACS Nano, 2010, 4(4), 2429–2437 CrossRef CAS PubMed.
- Z. Zhang, H. Chen, C. Xing, M. Guo, F. Xu, X. Wang, H. J. Gruber, B. Zhang and J. Tang, Nano Res., 2011, 4(6), 599–611 CrossRef CAS.
- J. Hossick-Schott, J. D. Norton and C. L. Schmidt, US Pat., US7002790, 2006 Search PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130(33), 10876–10877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details of spectroscopic, thermal stability and biocompatibility studies, microscopy images and electrochemical data. See DOI: 10.1039/c5cc00968e |
| This journal is © The Royal Society of Chemistry 2015 |