
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-infrared-induced electron transfer of an uranyl macrocyclic complex without energy transfer to dioxygen†
Christina M.
Davis
a,
Kei
Ohkubo
b,
I-Ting
Ho
a,
Zhan
Zhang
a,
Masatoshi
Ishida
c,
Yuanyuan
Fang
d,
Vincent M.
Lynch
a,
Karl M.
Kadish
*d,
Jonathan L.
Sessler
*a and
Shunichi
Fukuzumi
*be
aDepartment of Chemistry & Biochemistry, University Station-A5300, The University of Texas, Austin, Texas 78712-0165, USA. E-mail: sessler@cm.utexas.edu
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University and ALCA, Japan Science and Technology Agency (JST), 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
cEducation Centre for Global Leaders in Molecular Systems for Devices, Kyushu University, Fukuoka, 819-0395, Japan
dDepartment of Chemistry, and Department of Material and Life Science, University of Houston, Houston, Texas 77204-5003, Japan. E-mail: kkadish@uh.edu
eDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Korea
First published on 12th March 2015
Abstract
Photoexcitation of dichloromethane solutions of an uranyl macrocyclic complex with cyclo[1]furan[1]pyridine[4]-pyrrole (1) at the near-infrared (NIR) band (1177 nm) in the presence of electron donors and acceptors resulted in NIR-induced electron transfer without producing singlet oxygen via energy transfer.
For efficient light energy conversion, it is critical to harvest near-infrared (NIR)-range light, which constitutes nearly 50% of the solar energy.1 Extensive efforts have so far been made to synthesise chromophores which can absorb NIR light.2–18 However, to our knowledge, there is no chromophore that can absorb NIR light longer than 1000 nm and still act as a photosensitiser for NIR-induced electron transfer. This reflects the fact that the lifetimes of chromophores absorbing light longer than 1000 nm are typically too short to be quenched effectively by electron donors or acceptors.
Expanded porphyrins, large pyrrolic macrocycles with extended conjugation pathways, have attracted considerable attention in recent years due to their unique optical, electronic, chemical, and electrochemical properties.19–23 One salient feature of expanded porphyrins is the presence of Q-like bands with high extinction coefficients in the near-infrared (NIR) portion of the electromagnetic spectrum.24 We report here that the uranyl macrocyclic complex of cyclo[1]furan[1]pyridine[4]pyrrole (1; Fig. 1), which has an absorption maximum at 1177 nm,25 can undergo NIR-induced electron transfer via the triplet exited state with electron donors and acceptors without energy transfer to dioxygen to produce singlet oxygen. This finding is ascribed to the fact that both the one-electron oxidized radical cation and one-electron reduced radical anion forms of 1 are sufficiently stable to allow for characterisation by spectroscopic means.
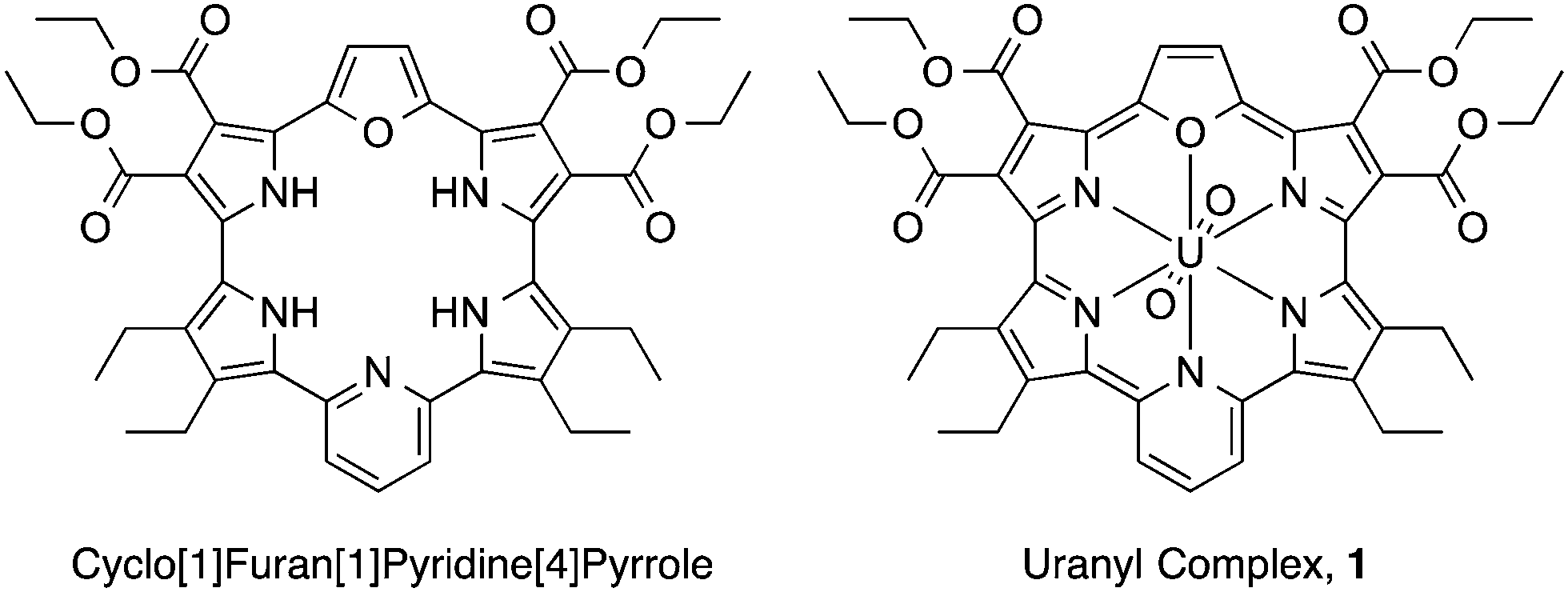 | ||
| Fig. 1 Cyclo[1]furan[1]pyridine[4]pyrrole and its uranyl complex (1). | ||
The uranyl macrocyclic complex (1) was synthesised as reported previously.25 Complex 1 has 22 π-electron aromatic heteroannulene character, exhibiting NIR absorption bands at 846 and 1177 nm as shown in Fig. 2 (black line). The one-electron oxidation of 1 with magic blue resulted in formation of the radical cation of 1 (1˙+), which has NIR absorption bands at 1280, 1732 and 1930 nm (Fig. 2, red and Fig. S1 in ESI†). These changes in the absorption were also seen under conditions of spectroelectrochemical oxidation at 0.76 V vs. SCE (Fig. S2 in ESI†).
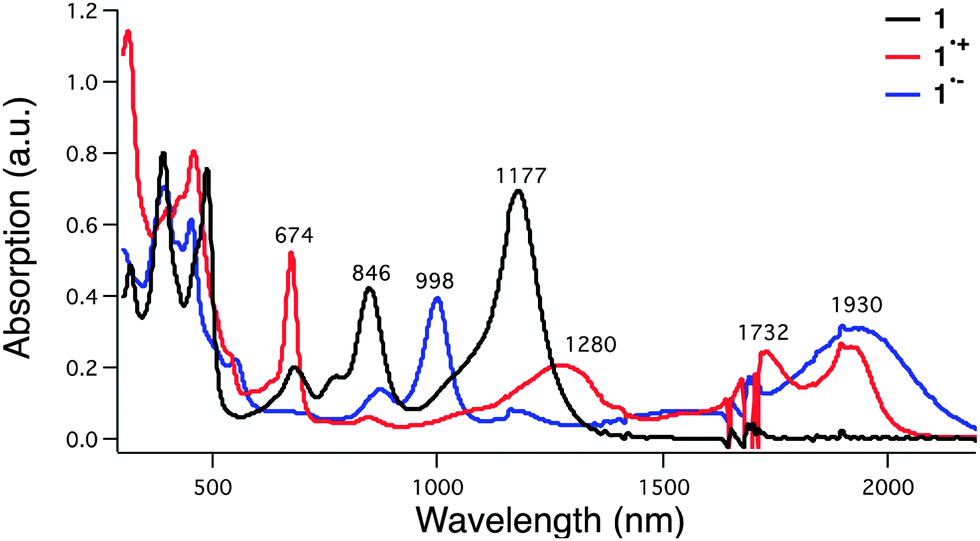 | ||
| Fig. 2 UV-Vis-NIR absorption spectra for 1 (black), 1 in the presence of 1 equivalent “magic blue” (1˙+, red), and 1 recorded in the presence of ∼1 equivalent cobaltocene (1˙−, blue) in dichloromethane at room temperature. [1] = 20 μM. | ||
A planar structure of the radical cation 1˙+ was obtained on the basis of an unrestricted DFT (UB3LYP/B1) calculation, which revealed spin delocalization over the macrocyclic core (Fig. S3b in ESI†). The singly occupied molecular orbitals (SOMOs) were found to be ligand-based MOs (Fig. S4 in ESI†). Time dependent (TD)-UDFT analyses of the radical cation 1˙+ revealed an allowed HOMO–LUMO transition band (f = 0.0513), a finding that is consistent with the experimental NIR absorption spectrum (Fig. S5 and Table S1 in ESI†). Thus, a stable open-shell [4n − 1] π-electron form is predicted under conditions of both chemical and electrochemical oxidation (see cyclic voltammogram in Fig. S6 in ESI†). In fact, the radical cation (1˙+) proved relatively stable with no significant change in the absorption spectrum over 50 minutes post addition of oxidant (Fig. S7 in ESI†).
1H-NMR spectroscopy was also used to monitor the production of the presumed radical cation. Adding aliquots of magic blue to a deuterated dichloromethane solution of 1 led a concentration dependent broadening of the peaks, as would be expected for the formation of a paramagnetic radical species (Fig. S8 in ESI†). A corresponding electron paramagnetic resonance (EPR) spectroscopic analyses of 1 revealed a signal at g = 2.0051 whose intensity increased upon the addition of magic blue until approximately 1.0 equiv. had been added (Fig. S9 in ESI†).
Slow diffusion of hexanes into a solution of dichloromethane containing 1 and one equiv. of magic blue led to the formation of diffraction grade single crystals. The crystal structure (Fig. 3) revealed that a β-position of one of the 2,5-diethyl substituted pyrrole rings is sp3 hybridized and bears a chlorine substituent, forming 1-Cl+·SbCl6−. When dissolved in dichloromethane, this crystalline product gives rise to an absorption spectrum more similar to that of neutral macrocycle 1 than the radical cation species 1˙+ (i.e., maxima at 846 and 1177 nm are seen, Fig. S10 in ESI†). Using the TD-DFT method, a simulated spectrum was generated that matches that of the structurally characterized chloride adduct to 12+ (see Fig. S11 in the ESI†). The intense NIR absorption feature can be interpreted in terms of broken degeneracy for the LUMO pairs due to the unsymmetric core structure (Fig. S12 and Table S2 in ESI†). The formation of the chloride adduct is ascribed to disproportionation of 1˙+, followed by the nucleophilic addition of Cl− to resulting dication, 12+, because the same absorption spectrum was obtained when complex 1 was oxidized with 1 equiv. of magic blue in the presence of ten equiv. tetrabutylammonium chloride (TBACl) for 10 min (Fig. S13 in ESI†). Once produced, the chloride adduct 1-Cl+ is stable in solution for at least 24 h (Fig. S14 in ESI†).
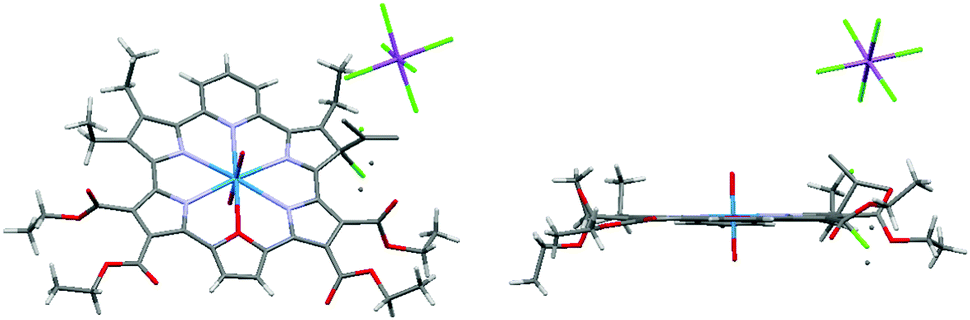 | ||
| Fig. 3 Top (left) side views (right) of the solid state structure of the chloride adduct of 1, 1-Cl·SbCl6. Crystals used for this analysis were obtained via the slow diffusion of hexanes into a dichloromethane solution of 1 containing 1 equiv. of “magic blue”. | ||
The radical anion 1˙− was produced by subjecting the original uranyl complex 1 to one-electron chemical reduction using cobaltocene as a reductant. The addition of cobaltocene to 1 resulted in the appearance of new absorption features at 998 and 1930 nm in the absorption spectrum (Fig. 2, blue and Fig. S15 in ESI†). These spectral features match those seen upon spectroelectrochemical reduction of 1 in dichloromethane (Fig. S16 in ESI†). An EPR spectrum expected for a radical (i.e., a feature at g = 2.003) was also recorded for this reduced species (Fig. S17 in ESI†). On this basis, it was assigned to be the 4n + 1 π-electron radical anion form of 1 (1˙−). The radical anion 1˙− proved stable for up to 16 hours in dichloromethane when produced via chemical reduction (Fig. S18 in ESI†). Similar spectral features were noted under conditions of spectroelectrochemistry when 1 was subject to reduction at −0.02 V vs. SCE (Fig. S16 in ESI†). Similarly, the spin delocalized structure was predicted for 1˙− (Fig. S19 in ESI†). A TD-DFT calculation produced a stick spectrum concordant with that of the experimental absorption spectrum for 1˙− (Fig. S20 in ESI†). The compositions of the major transitions of 1˙− are found to be straightforward; from any of the HOMOs to the LUMO (B) (Fig. S21 and Table S3 in ESI†). Furthermore, evaluation of the HOMOs for the radical cation and radical anion forms of 1 revealed increased conjugated character in the case of the radical anion 1˙− relative to the radical cation 1˙+. These differences may be reflected in the apparent greater stability observed for the radical anion (Table S4 in ESI†).
Femtosecond laser excitation of a CH2Cl2 solution of 1 at 393 nm resulted in observation of a transient absorption band at 620 nm with bleaching at 1150 nm, which matches with the absorption maximum of 1 as shown in Fig. 4.26 The time profile of the recovery of bleaching at 1150 nm could be fit to two-exponentials. The fast component has a lifetime of 2.5 ps (Fig. 4b), while the much slower component has a lifetime of 5 ns (Fig. 4c). The fast recovery component of the ground state bleaching at 1150 nm is ascribed to the relaxation from the singlet excited state to the ground state in competition with the fast intersystem crossing to form the triplet excited state due to the heavy atom effect of uranium.27 The much slower component is considered to reflect the decay of the triplet excited state of 1 (31*). The relatively short lifetime of 31* as compared with triplet excited states of organic compounds, which have microseconds lifetimes, reflects the large spin–orbit coupling of uranium.27
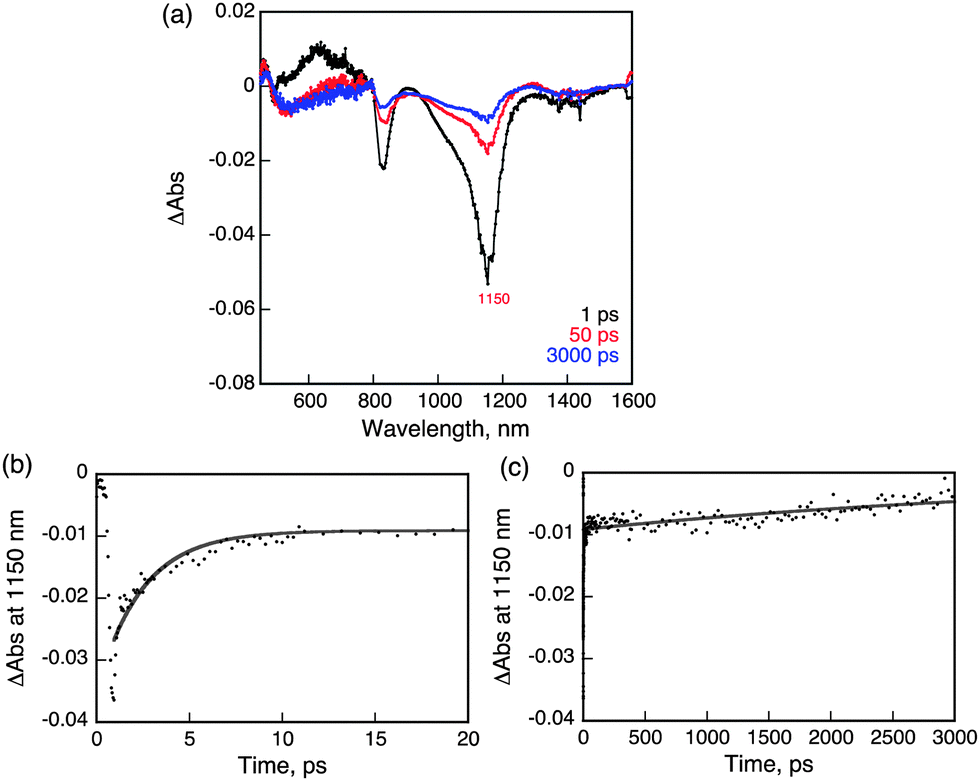 | ||
| Fig. 4 (a) Transient absorption spectra of 1 observed at 1, 50 and 3000 ps after femtosecond laser excitation of a deaerated CH2Cl2 solution of 1 (10 μM). (b) Time profile of bleaching at 1150 nm in the 0–20 ps delay time range. (c) Time profile of bleaching at 1150 nm in the 0–3000 ps delay time range. The gray lines in (b) and (c) are drawn by means of a double-exponential curve fitting. | ||
The ability of 31* to undergo reactions was examined first by whether it was able to induce the conversion of triplet oxygen (the ground state) to the corresponding singlet form. However, efforts to measure the phosphorescence due to singlet oxygen at 1273 nm in fully deuterated benzene (C6D6) revealed no appreciable signal. Additionally, we also confirmed that no oxygenation of anthracene, used as a singlet oxygen trap reagent, occurs (to produce epidioxyanthracene) in an O2-saturated CH2Cl2 solution containing 1 under visible-NIR photoirradiation with a solar light simulator attached with a light cut-off filter (λ > 490 nm, 1 sun for 3 h). Such an observation is consistent with the energy of 31* being lower than that of singlet oxygen (0.97 eV).28
In the presence of tetracyanoethylene (TCNE) as an electron acceptor, NIR-induced electron transfer from 31* to TCNE occurred as shown in Fig. 5a, where the negative transient absorption band due to the ground state bleaching at 1150 nm, which agree with NIR absorption band of 1 in Fig. 2, is recovered with a much faster rate in the presence of TCNE as compared with than that in its absence (Fig. 4c). The driving force of back electron transfer was calculated to be 0.54 eV based on the one-electron oxidation potential of 1 (0.76 V vs. SCE) and the one-electron reduction potential of TCNE (0.22 V vs. SCE),29 whereas the driving force of NIR-induced electron transfer is estimated to be smaller than 0.43 eV from the energy of 31* (<0.97 eV, vide supra). Because the driving force of back electron transfer is larger than that of NIR-induced electron transfer, back electron transfer may be faster than NIR-induced electron transfer thus accounting for the fact that no transient absorption features ascribable to 1˙+ or TCNE˙− were observed in Fig. 5a.30 The rate constant of electron transfer from 31* to TCNE was determined to be 6.7 × 109 M−1 s−1 from the slope of a linear plot of the pseudo-first-order rate constant of the NIR-induced electron transfer vs. [TCNE] (Fig. 5b).
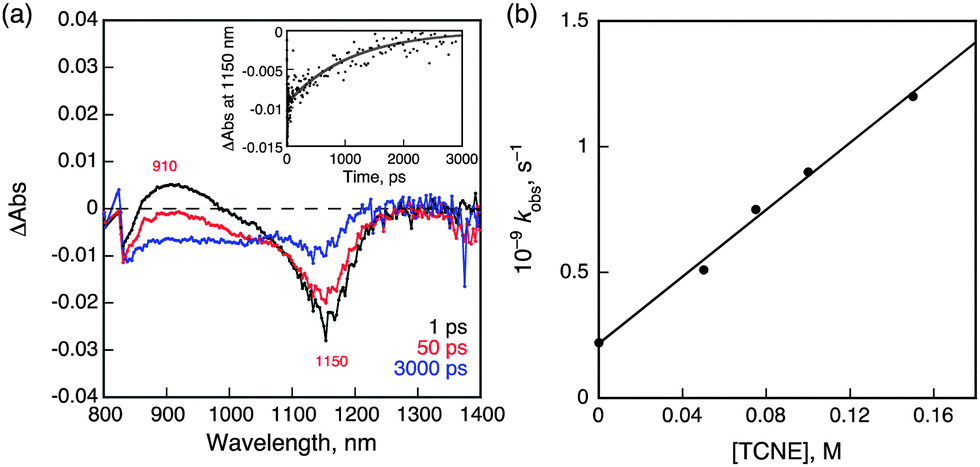 | ||
| Fig. 5 (a) Near-IR transient absorption spectra observed at 1, 50 and 3000 ps after femtosecond laser excitation of a deaerated CH2Cl2 solution of 1 (50 μM) and TCNE (100 mM). Inset: time profile of the absorbance at 1150 nm. (b) Plot of kobsvs. [TCNE]. | ||
When TCNE was replaced by 9,10-dihydro-10-methylacridine (AcrH2) as an electron donor, NIR-induced electron transfer from AcrH2 to 31* occurred as shown in Fig. 6. The driving force of back electron transfer is determined to be 0.83 eV from the one-electron oxidation potential of AcrH2 (0.81 V vs. SCE)30 and the one-electron reduction potential of 1 (−0.02 V vs. SCE), whereas the driving force of NIR-induced electron transfer is estimated to be smaller than 0.14 eV from the energy of 31* (<0.97 eV). In this case as well, back electron transfer from 1˙− to AcrH2˙+ was faster than the NIR-induced electron transfer because of the larger driving force of back electron transfer at 298 K. The rate constant of electron transfer from AcrH2 to 31* was determined to be 9.0 × 109 M−1 s−1 form the slope of a linear plot of the pseudo-first-order rate constant of NIR-induced electron transfer vs. concentration of AcrH2 (Fig. 6b).30 This value is close to the diffusion limited in CH2Cl2 (∼1010 M−1 s−1). On this basis, we conclude that electron transfer from AcrH2 to 1 is an exergonic process. The radical ion pair was detected by EPR spectroscopy under NIR photoirradiation (λ > 490 nm) at low temperature (77 K). The EPR spectrum is characterized by a broad signal due to AcrH2˙+,30 which overlaps with the signal due to 1˙− at g = 2.003 (see ESI,† Fig. S22).
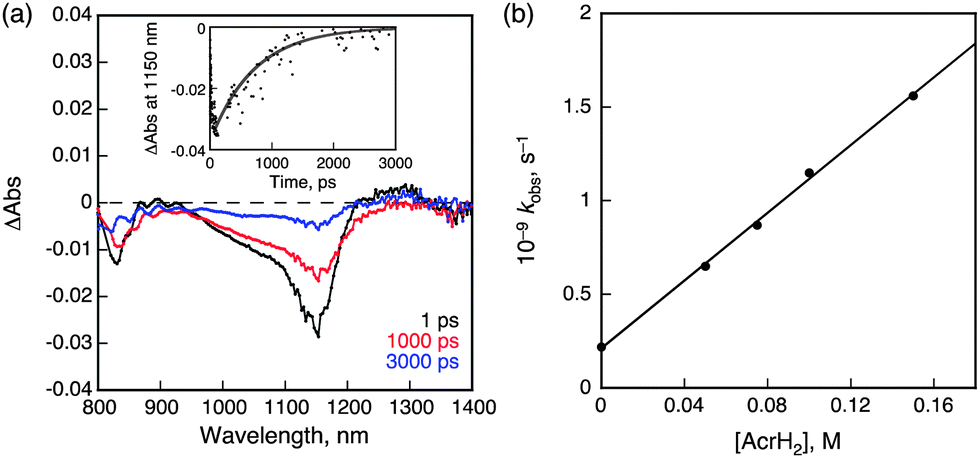 | ||
| Fig. 6 (a) Near-IR transient absorption spectra observed at 1, 1000, and 3000 ps after femtosecond laser excitation of a deaerated CH2Cl2 solution of 1 (50 μM) and AcrH2 (100 mM). Inset: time profile of the absorbance at 1150 nm; (b) plot of kobsvs. [AcrH2]. | ||
In conclusion, the uranyl macrocyclic complex (1) can be converted readily to the corresponding radical cation and radical anion states, both of which are stable on the laboratory timescale. The ability to access both 4n + 1 and 4n − 1 π-electron states is thought to underlie the fact that complex 1 acts as an efficient NIR-absorbing photosensitiser with a low triplet excited state energy (<0.97 eV). Specifically, it undergoes NIR-induced electron transfer with appropriately chosen electron donors and acceptors without energy transfer to dioxygen. Such a NIR photosensitiser, characterized by its absorption maximum at 1177 nm, may find applications for NIR light energy conversion. Likewise, the ability to stabilize multiple electronic states may allow for further insights into odd electron species that lie formally between closed shell aromatic 4n + 2 and antiaromatic 4n π-electron states.
This work was supported by ALCA and SENTAN programs from Japan Science Technology Agency (JST) (to S.F.), Japan, Grant-in-Aid (No. 26620154 and 26288037) from MEXT, Japan (to K.O.), a U.S. NSF grant (CHE 1057904 to J.L.S.), and the Robert A. Welch Foundation (grants F-1018 and E-680 to J.L.S. and K.M.K.)
Notes and references
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- H. Rath, A. Mallick, T. Ghosha and A. Kalita, Chem. Commun., 2014, 50, 9094 RSC.
- S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed.
- V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812 CrossRef CAS PubMed.
- L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119 CrossRef CAS PubMed.
- S. Ishida, T. Tanaka, J. M. Lim, D. Kim and A. Osuka, Chem. – Eur. J., 2014, 20, 8274 CrossRef CAS PubMed.
- Y. Wang, J. Chen, Y. Zhen, H. Jiang, G. Yu, Y. Liu, E. Baranoff, H. Tan and W. Zhu, Mater. Lett., 2015, 139, 130 CrossRef CAS PubMed.
- K. Naoda, H. Mori and A. Osuka, Chem. – Asian J., 2013, 8, 1395 CrossRef CAS PubMed.
- H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS PubMed.
- S. K. Sarkar, S. Mukherjee and P. Thilagar, Inorg. Chem., 2014, 53, 2343 CrossRef CAS PubMed.
- G. Li, A. Yella, D. G. Brown, S. I. Gorelsky, M. K. Nazeeruddin, M. Grätzel, C. P. Berlinguette and M. Shatruk, Inorg. Chem., 2014, 53, 5417 CrossRef CAS PubMed.
- K. C. D. Robson, B. D. Koivisto, A. Yella, B. Sporinova, M. K. Nazeeruddin, T. Baumgartner, M. Grätzel and C. P. Berlinguette, Inorg. Chem., 2011, 50, 5494 CrossRef CAS PubMed.
- C.-J. Yao, Y.-W. Zhong and J. Yao, J. Am. Chem. Soc., 2011, 133, 15697 CrossRef CAS PubMed.
- W.-W. Yang, J. Yao and Y.-W. Zhong, Organometallics, 2012, 31, 8577 CrossRef CAS.
- H.-J. Nie and Y.-W. Zhong, Inorg. Chem., 2014, 53, 11316 CrossRef CAS PubMed.
- C. Liu, M. P. Dobhal, M. Ethirajan, J. R. Missert, R. K. Pandey, S. Balasubramanian, D. K. Sukumaran, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 14311 CrossRef CAS PubMed.
- A. Kozyrev, M. Ethirajan, P. Chen, K. Ohkubo, B. C. Robinson, K. M. Barkigia, S. Fukuzumi, K. M. Kadish and R. K. Pandey, J. Org. Chem., 2012, 77, 10260 CrossRef CAS PubMed.
- A. Jasat and D. Dolphin, Chem. Rev., 1997, 97, 2267 CrossRef CAS PubMed.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed.
- M. Ishida, J.-Y. Shin, J. M. Lim, B. S. Lee, M.-C. Yoon, T. Koide, J. L. Sessler, A. Osuka and D. Kim, J. Am. Chem. Soc., 2011, 133, 15533 CrossRef CAS PubMed.
- H. Mori, T. Tanaka and A. Osuka, J. Mater. Chem. C, 2013, 1, 2500 RSC.
- T. Yoneda and A. Osuka, Chem. – Eur. J., 2013, 19, 7314–7318 CrossRef CAS PubMed.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- I.-T. Ho, Z. Zhang, M. Ishida, V. M. Lynch, W.-Y. Cha, Y. M. Sung, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 4281 CrossRef CAS PubMed.
- O. Bilsel, S. N. Milam, G. S. Girolami, K. S. Suslick and D. Holten, J. Phys. Chem., 1993, 97, 7216 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685 CrossRef CAS PubMed.
- S. Fukuzumi, C. L. Wong and J. K. Kochi, J. Am. Chem. Soc., 1980, 102, 2928 CrossRef CAS.
- We could not detect the radical ion pair spectral features corresponding to the electron-transfer products via transient absorption spectroscopy. We rationalize this finding in terms of the rate of back electron transfer being faster than that of photoinduced electron transfer at 298 K.
- S. Fukuzumi, Y. Tokuda, T. Kitano, T. Okamoto and J. Otera, J. Am. Chem. Soc., 1993, 115, 8960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic details. CCDC 1045373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00903k |
| This journal is © The Royal Society of Chemistry 2015 |