
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic one-electron reduction of uranyl(VI) to Group 1 uranyl(V) complexes via Al(III) coordination†
Markus
Zegke
,
Gary S.
Nichol
,
Polly L.
Arnold
* and
Jason B.
Love
*
EaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: Polly.Arnold@ed.ac.uk; Jason.Love@ed.ac.uk; Fax: +44 (0) 130 650 6453; Tel: +44 (0) 130 650 5429
First published on 2nd March 2015
Abstract
Reactions between the uranyl(VI) Pacman complex [(UO2)(py)(H2L)] of the Schiff-base polypyrrolic macrocycle L and Tebbe's reagent or DIBAL result in the first selective reductive functionalisation of the uranyl oxo by Al to form [(py)(R2AlOUO)(py)(H2L)] (R = Me or iBu). The clean displacement of the oxo-coordinated Al(III) by Group 1 cations has enabled the development of a one-pot, DIBAL-catalysed reduction of the U(VI) uranyl complexes to a series of new, mono-oxo alkali-metal-functionalised uranyl(V) complexes [(py)3(MOUO)(py)(H2L)] (M = Li, Na, K).
The uranyl(VI) dication UO22+ is the most common form of uranium in the environment, and is reduced by minerals and microbes to the less stable uranyl(V) monocation UO2+, the chemistry of which has only recently been investigated in detail.1–4 The [Rn] 5f1-electron configuration of uranyl(V) results in a variety of interesting properties such as cation–cation interactions (CCIs)5 and single molecule magnetism (SMM),6 and provides insight into often non-trivial 5f-electron behaviours.7 Exploring the uranyl(V) oxidation state may help in understanding the fundamental uranium-based processes occurring in groundwater remediation and nuclear fuel corrosion.8 Furthermore, due to the increased Lewis basicity of UVO2+ compared to UVIO22+,9 uranyl(V) complexes may also be employed to model the behaviour of highly radioactive neptunyl ions NpO22+ which are present in nuclear waste.10 Studies by us,11 and others,12,13 on the reactions of uranyl(VI) complexes with silyl-containing reagents have led to new reductive oxo-functionalisation reactions being uncovered, forming stable silylated uranyl(V) complexes and a chemically inert and air-stable butterfly-shaped bimetallic uranium(V) dioxo complex.14 As with reductive metalation reactions of the uranyl(VI) dication, the stability of uranyl(V) complexes against disproportionation is dramatically enhanced through the functionalisation of the more Lewis-basic oxo group of this f1 cation.15,16 Here, we report synthetic routes to the first oxo-aluminated uranyl(V) complexes, transmetalation reactions to the first mono-alkali metal uranyl(V) adducts supported by the Pacman ligand, and a new procedure to alkali-metal functionalised uranyl(V) complexes that is catalytic in the Al(III) reagent. Significantly, these complexes are exclusively exo-oxo metalated, and show high stability against disproportionation to uranyl(VI) and uranium(IV) compounds.17,18
We have studied a range of AlIII compounds that might behave as suitable electrophiles to the accessible oxo group of the uranyl ion in the uranyl(VI) Pacman complex [(UO2)(py)(H2L)] A.19 In particular, two compounds [Cp2Ti(μ-Cl)(μ-CH2)AlMe2] (Tebbe's reagent) and [(iBu)2AlH]2 (DIBAL) have proven to be excellent sources of the oxophilic and Lewis acidic AlIII cation (Scheme 1).
 | ||
| Scheme 1 Reductive alumination of [(UO2)(py)(H2L)], A by Tebbe's reagent or DIBAL. | ||
The combination of benzene solutions of equimolar quantities of A and [Cp2Ti(μ-Cl)(μ-CH2)AlMe2], followed by the addition of 0.1 mL of pyridine at room temperature results in a clear orange solution from which yellow crystals form upon standing, characterised as [(py)(Me2AlOUO)(py)(H2L)] 1, and isolated in 67% yield. The X-ray crystal structure of 1 was determined and shows the expected wedge-shaped Pacman geometry of the parent complex with exo-oxo aluminium coordination (Fig. 1). The uranyl oxo groups adopt a trans geometry, with an O1–U1–O2 angle of 174.3(1)° and U1–O1 and U1–O2 bond lengths elongated to 1.857(3) Å and 1.962(3) Å respectively, compared to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO bonds of 1.793(6) Å and 1.773(6) Å for A.19 This significant lengthening of these bonds is indicative of a decrease in the uranyl bond order and is similar to related experimental and calculated systems in which an increase of 0.151–0.242 Å in U–O bond lengths upon reduction of OUVIO to OUV–O–M is seen.20 Furthermore, the hydrogen-bonding interactions between the endo-oxo O1 and the two pyrrole protons in the vacant macrocyclic pocket, shown by O1⋯N1 2.964(5) Å and O1⋯N2 3.068(5) Å are slightly shorter than those in A (3.111(7) Å and 3.146(7) Å) and supports the enhanced oxo basicity of the f1 cation. To our knowledge, this is the first reaction in which Tebbe's reagent is used as a source of aluminium.
UO bonds of 1.793(6) Å and 1.773(6) Å for A.19 This significant lengthening of these bonds is indicative of a decrease in the uranyl bond order and is similar to related experimental and calculated systems in which an increase of 0.151–0.242 Å in U–O bond lengths upon reduction of OUVIO to OUV–O–M is seen.20 Furthermore, the hydrogen-bonding interactions between the endo-oxo O1 and the two pyrrole protons in the vacant macrocyclic pocket, shown by O1⋯N1 2.964(5) Å and O1⋯N2 3.068(5) Å are slightly shorter than those in A (3.111(7) Å and 3.146(7) Å) and supports the enhanced oxo basicity of the f1 cation. To our knowledge, this is the first reaction in which Tebbe's reagent is used as a source of aluminium.
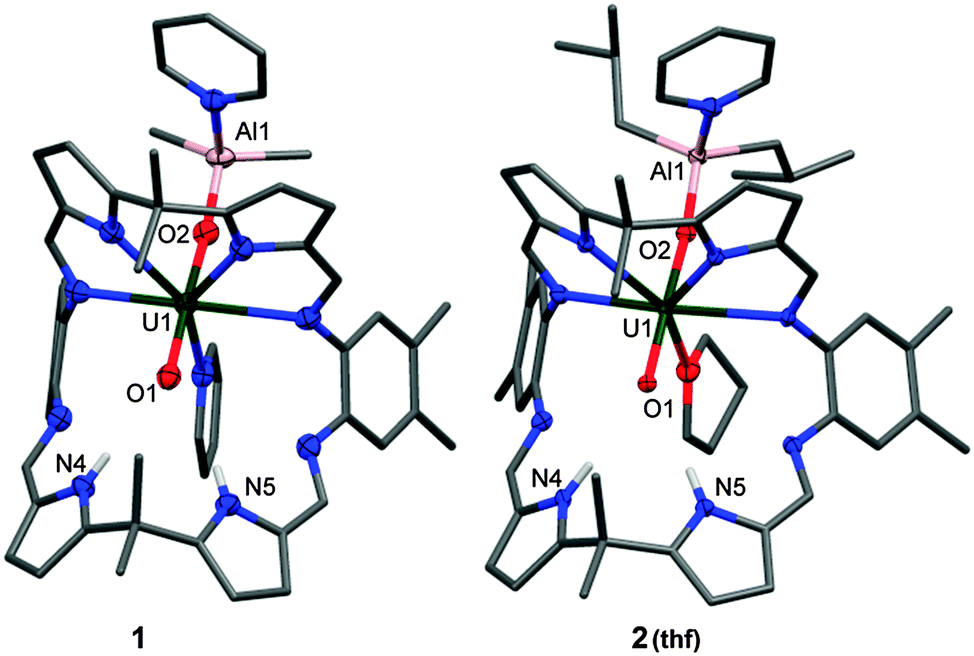 | ||
| Fig. 1 Solid-state structures of 1 and 2(thf), front view. For clarity, all hydrogen atoms except the pyrrole NHs and all solvent molecules are omitted (displacement ellipsoids are drawn at 50% probability). Selected bond lengths (Å) and angles (°) for 1: U1–O1 1.857(3), U1–O2 1.962(3), O1–N4 2.964(5), O1–N5 3.068(5). O1–U1–O2 174.3(1); 2: U1–O1 1.855(2), U1–O2 1.962(2), O1–N4 3.033(4), O1–N5 3.027(4). O1–U1–O2 175.1(1). | ||
A more atom-economic route to these heterobimetallic complexes is through the reaction between A and DIBAL in toluene at 70 °C for 24 h which results in the formation of yellow [(py)(iBu2AlOUO)(py)(H2L)] 2 in 51% yield. The solid state structure of 2 (Fig. 1) is very similar to 1, once more exemplifying the formal UV oxidation state through an elongation of the U1–O1 and U1–O2 bond distances. Mechanistically, it is likely that 1 and 2 are formed through Al–ligand bond homolysis (Al–H or Al–C) which provides the reducing electron. This process is similar to that suggested by us previously to be responsible for UVI reduction in the formation of lithium-functionalised [(LiOUO)(py)(Li3L)] and lanthanide-functionalised [UO2(py)(Ln(py)L)]2 that result from Li–C or Ln–N bond homolysis.21
The 1H-NMR spectrum of 2 (see ESI†) shows contact-shifted and broadened resonances between −6 and +70 ppm due to the paramagnetism of the f1 centre. Even so, the iBu methyl hydrogens can be identified at 6.10 ppm and 6.67 ppm with 3JH–H coupling of 8 Hz, and the methine proton is a broad resonance at 11.31 ppm that couples with the methylene protons at 16.35 ppm and 16.81 ppm. The most contact-shifted resonance at +69 ppm is assigned to the pyrrole N–H protons. In situ measurements show the formation of gaseous H2 at 4.49 ppm. Both UVO2–AlIII compounds 1 and 2 are stable in THF and pyridine solvents. A study of the redox chemistry of 2 by cyclic voltammetry in THF with 0.1 M [NBu4][PF6] as a supporting electrolyte at 500 mV s−1 reveals a quasi-reversible reduction at E1/2 = –1.42 V (vs. Fc/Fc+) which is tentatively ascribed to a uranyl(V)/uranium(IV) redox couple (see ESI†). A pre-reduction wave is also seen at Epc = −1.45 V, implying that the redox chemistry of 1 and 2 is not straightforward, and as yet, in line with related U(V) complexes that we have studied, the chemical reduction of complex 1 or 2 has not yet been successful. However, we have found that the AlR2 group is readily substituted by Group 1 metal cations by reaction with an alkyl or hydride reagent such as MeLi, NaH or KH (Scheme 2); these experiments had been anticipated to deprotonate the two, likely acidic, pyrrole NHs in 1 and 2.
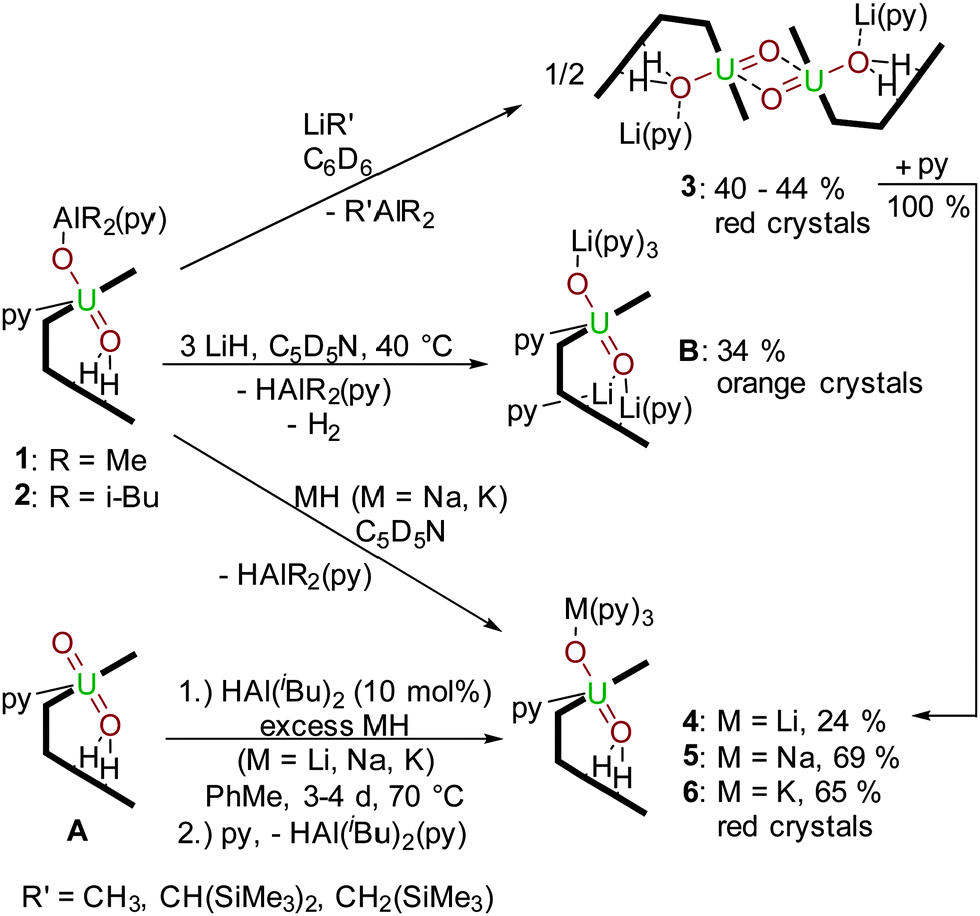 | ||
| Scheme 2 Transmetalation reactions of 1 and 2 with R′Li, LiH, NaH and KH, and the HAl(i-Bu)2-catalysed reduction of [UVIO2(H2L)] by MH. | ||
Reactions between benzene solutions of 1 with either one or two equivalents of the strong base MeLi affords solely [{(OUO)Li(py)(H2L)}2] 3 in moderate isolated yield (40%), which remains UV and doubly NH protonated. This contrasts with the reactions of the uranyl(VI) Pacman complex [UO2(py)(H2L)] A with single equivalents of LiR (R = H, NH2, NiPr2, N(SiMe3)2, CPh3, C5H5) that simply result in pyrrole deprotonation to afford the uranyl(VI) complex [(UO2)(py)(LiHL)],22 and suggests that the hydrogen-bonding interaction between the f1 uranyl oxo group and the pyrrole protons is significant enough to attenuate deprotonation. The X-ray crystal structure of 3 (Fig. 2) shows that the lithium cation is coordinated by the imine groups of the macrocycle and that the uranium centre has migrated from its usual N4 donor pocket to an alternative pyrrole–imine–imine–pyrrole set. This results in the macrocycle folding at the meso-carbons and not the aryl groups, so resulting in a ‘bowl-shaped’ geometry.23–25
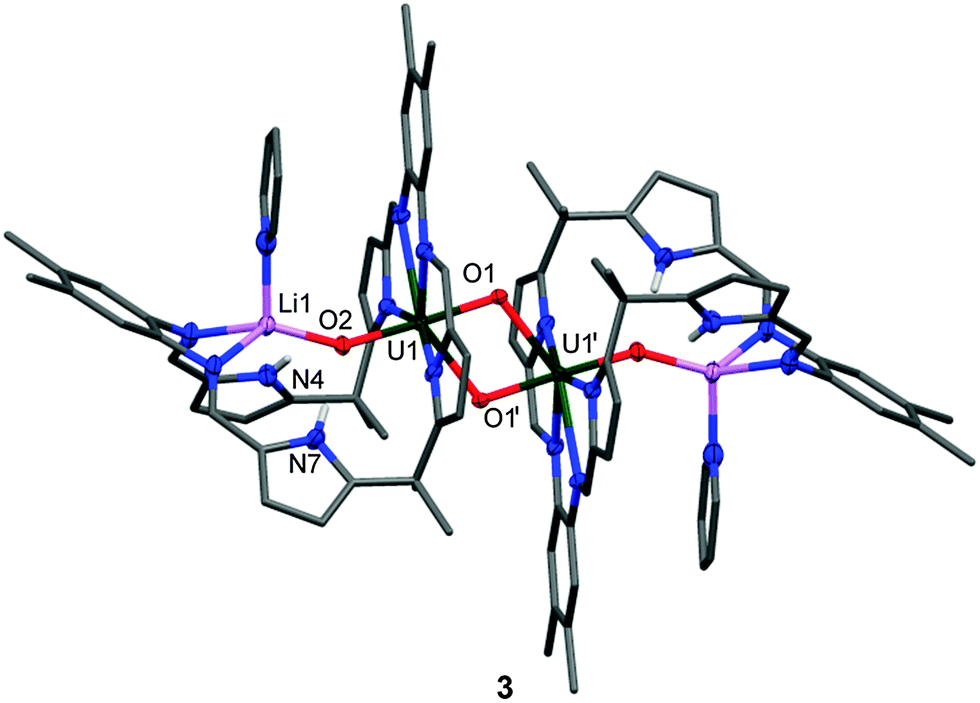 | ||
| Fig. 2 Solid-state structure of 3. For clarity, all hydrogen atoms except pyrrole NH and all solvent molecules are omitted (displacement ellipsoids are drawn at 50% probability). Selected bond lengths (Å) and angle (°): U1–O1 1.908(2), U1–O2 1.891(2), U1–O1′ 2.372(3), N4–O2 3.269(4), N7–O2 3.198(5). O1–U1–O2 177.7(1). U1⋯U1′: 3.5199(9). | ||
As above, the U1–O1 and U1–O2 bond lengths of 1.891(2) Å and 1.908(2) Å, respectively are elongated compared to those in A, supporting a UV oxidation state, and the oxo groups are arranged in a trans disposition. The Li cation is thus sited within the cavity of the macrocycle, bound to the uranyl endo-oxo atom, the two imine groups, and a molecule of pyridine. As in the other complexes the uranyl is five-coordinate in the equatorial plane but the site which was occupied by the donor solvent is now filled by the exo-oxo group of its counterpart in the dimer, resulting in a diamond-shaped U2O4–cation–cation interaction.1,2,16 The uranium–uranium separation in this dimer is short at 3.5199(9) Å, but similar to other complexes previously reported by us, for example 3.4487(4) Å in the uranyl(V) yttrium dimer [{UO2Y(py)2(L)}2].26 Treatment of 2 with one equivalent of LiCH2SiMe3 or LiCH(SiMe3)2 in benzene also yielded 3, and was verified by 1H-NMR spectroscopy and crystal structure analysis (unit cell check). In contrast, reactions between 1 or 2 and an excess of LiH in the donor solvent pyridine at 40 °C results in the formation of the known, triply lithiated, uranyl(V) complex [(py)3(LiOUO)(py)({Li(py)}2L)] B.21
The difference in ability of the two types of Li reagents (LiR vs. LiH) to effect N–H deprotonation is likely due to the nature of the reaction solvent. The use of pyridine stabilises the exogenous coordination of the Li cation to the uranyl oxo group, whereas in benzene, the reorganization of the uranyl coordination pocket allows for maximum interaction of the Li cation with the macrocycle. In support of this, the addition of pyridine to a benzene solution of 3 shows a rearrangement from the bowl-shaped structure to 4, possessing the classical Pacman structure (Fig. 3), suggesting that the bowl-shaped structure is only favoured in a non-coordinating solvent. Exogenous coordination of a Li cation was also seen in the cleavage products of the heterobimetallic lanthanide-uranyl(V) dimers [{(UO2)Ln(py)2(L)}2] (Ln = Sc, Y, Ce, Sm, Eu, Gd, Dy, Er, Yb, Lu) to yield [(py)3(LiOUO)Ln(py)2(L)].27
 | ||
| Fig. 3 Solid-state structures of 4, 5 and 6 (side view). For clarity, all hydrogen atoms except pyrrole NHs and all solvent molecules are omitted (displacement ellipsoids are drawn at 50% probability). Selected bond lengths (Å) and angles (°) for 4: U1–O1 1.853(6), U1–O2 1.884(7), O1–N4 3.09(1), O1–N5 3.10(1). O1–U1–O2 173.8(3); 5: U1–O1 1.844(5), U1–O2 1.856(7), O1–N4 3.010(9), O1–N5 2.988(8). O1–U1–O2 174.2(3); 6: U1–O1 1.871(2), U1–O2 1.837(2), O1–N4 2.898(4), O1–N5 2.932(4). O1–U1–O2 176.1(1). | ||
Treatment of 1 or 2 with NaH or KH in pyridine at room temperature results exclusively in the exchange of the aluminium cation for the respective alkali metal to yield [(py)3(NaOUO)(py)(H2L)] 5 and [(py)3(KOUO)(py)(H2L)] 6 (Na: 69%, K: 65%); in both of these cases, the lower pocket remains protonated (Fig. 3). This is confirmed by the 1H-NMR resonances of 4, 5 and 6 in deuterated pyridine of which the pyrrole N–Hs show the most significant shifts to high frequencies at 85.48 ppm (4), 91.11 ppm (5) and 93.06 (6). Additionally the 7Li-NMR of 4 shows only one resonance at 88.48 ppm, supporting Li coordination to the paramagnet.
The solid state structures of 4, 5 and 6 are very similar, and in contrast to 3 show the classic uranyl Pacman geometry. The main difference between the structures is that the U1–O2–M1 angle is nearly linear for the Li (4) (173.8(3)°) and Na (5) (174.7(6)°) complexes whereas the U1–O2–K1 angle is considerably bent (116.0(1)°). This is caused by an η5-interaction between K and a U-bound pyrrolide ring due to the softness and size of K+ (152 pm) compared to Na+ (112 pm) and Li+ (73 pm).28
It is clear from the above transmetalation reactions that the aluminium by-product is the alane or aluminium hydride. As such it was envisaged that formation of the reduced, alkali-metalated uranyl complexes 4–6 from A should be achievable using MH (M = Li, Na, K) and a catalytic amount of AlH(iBu)2, as this latter reagent should be regenerated during the transmetalation step. As such, reactions using 10 mol% of DIBAL and an excess of MH in toluene at 70 °C for 72 to 96 hours (Scheme 2) were carried out and are found to generate 4, 5 or 6 in essentially quantitative yields (Table 1, showing reactions using KH only). Control reactions with no aluminium reagent formed 50% of 6 after 96 hours, and increasing the reaction time up to ten days afforded a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 6 and A; as such, reactions that incorporate DIBAL are significantly accelerated. Treatment of A with 5 mol% of DIBAL only gave 20% of 6 with 80% of the starting material still present, even after a prolonged reaction time.
:1 mixture of 6 and A; as such, reactions that incorporate DIBAL are significantly accelerated. Treatment of A with 5 mol% of DIBAL only gave 20% of 6 with 80% of the starting material still present, even after a prolonged reaction time.
| Entry | mol% HAl(i-Bu)2 | Time/h | Ratio 6/A |
|---|---|---|---|
| Reaction conditions: 70 °C, toluene, 5 equivalents KH. | |||
| 1 | 5 | 24 | 20/80 |
| 2 | 5 | 60 | 20/80 |
| 3 | 10 | 96 | 100/0 |
| 4 | 0 | 96 | 50/50 |
| 5 | 0 | 240 | 80/20 |
Additionally the redox chemistry of 6 was studied by cyclic voltammetry in THF with 0.2 M [NBu4][PF6] as a supporting electrolyte at scan rates between 100 and 500 mV s−1 and reveals a quasi-reversible reduction at E1/2 = –1.31 V (vs. Fc/Fc+) which is ascribed to a uranyl(V)/uranyl(VI) redox couple (see ESI†).
We report the first reductive alumination of the uranyl dication which results in a significant attenuation of the acidity of the pyrrole NHs through hydrogen bonding to the f1 centre, such that reactions with Group 1 bases result in transmetalation instead of the deprotonation chemistry previously seen. This change in reactivity has allowed us to develop a new synthetic route to simple, Group 1 cation adducts of uranyl(V) Pacman complexes that is catalytic in aluminium reagent. This new Al-mediated route should provide opportunities for new catalysed uranyl functionalisation reactions with other d- and f-group metal cations, and could even offer a general low-cost, one-pot route to the selective Group-1 cation metalation of d-block metal oxo complexes.
The authors thank the EPSRC-UK, the University of Edinburgh, the EU Actinet-I3-AC3-JRP-02 and Talisman-JRPL-C02-07 programs, COST CM1006, and the Institute of Advanced Study, TU Munich for support. MZ thanks Dr Stephen M. Mansell for valuable advice.
Notes and references
- P. L. Arnold, G. M. Jones, Q.-J. Pan, G. Schreckenbach and J. B. Love, Dalton Trans., 2012, 41, 6595–6597 RSC.
- P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS PubMed.
- D. D. Schnaars, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 9642–9649 CrossRef CAS PubMed.
- G. Nocton, P. Horeglad, V. Vetere, J. Pécaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef CAS PubMed.
- V. Mougel, J. Pécaut and M. Mazzanti, Chem. Commun., 2012, 48, 868 RSC.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- T. W. Hayton and G. Wu, Inorg. Chem., 2009, 48, 3065–3072 CrossRef CAS PubMed.
- P. C. Burns, R. C. Ewing and A. Navrotsky, Science, 2012, 335, 1184–1188 CrossRef CAS PubMed.
- J.-C. Berthet, G. Siffredi, P. Thuéry and M. Ephritikhine, Chem. Commun., 2006, 3184–3186 RSC.
- R. Copping, V. Mougel, C. Den Auwer, C. Berthon, P. Moisy and M. Mazzanti, Dalton Trans., 2012, 41, 10900–10902 RSC.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–317 CrossRef CAS PubMed.
- J. L. Brown, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 7248–7249 CrossRef CAS PubMed.
- J.-C. Berthet, G. Siffredi, P. Thuéry and M. Ephritikhine, Eur. J. Inorg. Chem., 2007, 4017–4020 CrossRef CAS.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- G. M. Jones, P. L. Arnold and J. B. Love, Angew. Chem., Int. Ed., 2012, 51, 12584–12587 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 9456–9458 CrossRef CAS PubMed.
- A. Ekstrom, Inorg. Chem., 1974, 13(9), 2237–2241 CrossRef CAS.
- H. Steele and R. J. Taylor, Inorg. Chem., 2007, 46, 6311–6318 CrossRef CAS PubMed.
- P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 CrossRef CAS PubMed.
- S. O. Odoh and G. Schreckenbach, Inorg. Chem., 2013, 52, 245–257 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 CrossRef CAS.
- J. W. Leeland, F. J. White and J. B. Love, J. Am. Chem. Soc., 2011, 133, 7320–7323 CrossRef CAS PubMed.
- J. B. Love, Chem. Commun., 2009, 3154 RSC.
- G. Givaja, M. Volpe, J. W. Leeland, M. A. Edwards, T. K. Young, S. B. Darby, S. D. Reid, A. J. Blake, C. Wilson, J. Wolowska, E. J. L. McInnes, M. Schröder and J. B. Love, Chem. – Eur. J., 2007, 13, 3707–3723 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J.-C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and characterising data. CCDC 1046279–1046284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00867k |
| This journal is © The Royal Society of Chemistry 2015 |