
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSirtuin inhibitor sirtinol is an intracellular iron chelator†
R.
Gautam
,
E. A.
Akam
,
A. V.
Astashkin
,
J. J.
Loughrey
and
E.
Tomat
*
University of Arizona, Department of Chemistry and Biochemistry, 1306 E. University Blvd., Tucson AZ 85721, USA. E-mail: tomat@email.arizona.edu
First published on 25th February 2015
Abstract
Sirtinol is a known inhibitor of sirtuin proteins, a family of deacetylases involved in the pathophysiology of aging. Spectroscopic and structural data reveal that this compound is also an iron chelator forming high-spin ferric species in vitro and in cultured leukemia cells. Interactions with the highly regulated iron pool therefore contribute to its overall intracellular agenda.
The architectures of biologically active small molecules, including pharmaceuticals,1 feature multiple heteroatoms (e.g., O, N, S) that can interact with metal ions. The ability of such compounds to recruit metal cations in the biological milieu could not only modify on-target efficacy, but also affect intracellular metal homeostasis and introduce new reactivity (for instance, through the redox chemistry of metal complexes formed in situ).2 These effects could lead to the repositioning of known drugs and/or to the identification of new therapeutic opportunities. Therefore, the potential for biological effects and/or polypharmacologic profiles involving metal coordination should be evaluated carefully. This study illustrates the iron binding properties of an established inhibitor of sirtuin proteins, sirtinol, which features a 2-hydroxiphenyl imine motif analogous to the core structure of several 2-hydroxyphenyl hydrazone chelators of transition metals.
Sirtuin proteins are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases present in mammalian cells in seven isoforms (SIRT1-7).3 Because of their role in histone modification and gene regulation, the study of sirtuins is relevant to the understanding (at a molecular level) of aging and age-related diseases such as neurodegenerative disorders, diabetes, and cancer.4,5 These implications have placed sirtuins at the center of intense investigation in the pharmaceutical arena in recent years. The activators6 and inhibitors7,8 identified over the last decade are the molecular tools employed to unravel the roles of sirtuin proteins, ranging from lifespan-extending effects to cell survival and metabolic control, to inflammatory response. The information collected through these investigations will ultimately determine the effective potential of sirtuins as therapeutic targets.
Sirtinol (Chart 1) is a member of the first cohort of sirtuin inhibitors identified by phenotypic screening.9 This inhibitor of SIRT1 and SIRT2 has been employed in multiple studies aimed at establishing sirtuins as therapeutic targets in anticancer drug development.10–16 Although specificity of protein interaction is the ideal scenario in such investigations, the occurrence of off-target effects is often difficult to rule out. In the case of sirtinol, the observation of biological activity at concentrations lower than the inhibitory levels (<25 μM) has suggested its effects on multiple intracellular pathways.17 Based on a structural analysis of its molecular scaffold, we sought to investigate the effect of sirtinol on intracellular iron homeostasis.
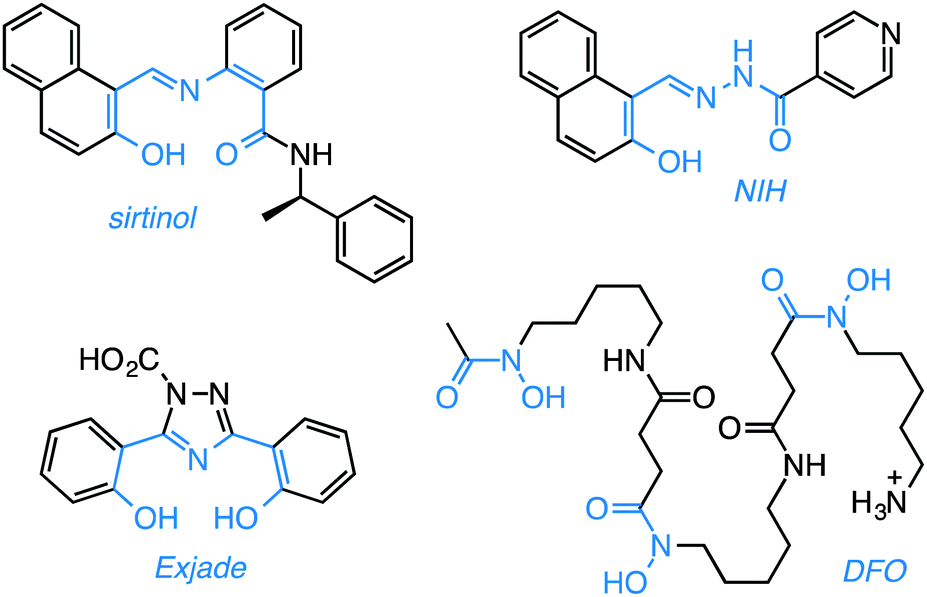 | ||
| Chart 1 Sirtuin inhibitor sirtinol and selected biologically-active iron chelators. The metal-binding units are highlighted in blue. | ||
From a structural standpoint, sirtinol shares several characteristics of effective metal-coordinating species. Within its scaffold, a 2-hydroxynaphthalenyl moiety is connected to a benzamide through an aldiminic nitrogen atom. Sirtinol thus presents a tridentate O,N,O donor set (Chart 1) similar to the binding units of several established iron scavengers, e.g., deferasirox (Exjade),18 a clinically approved chelator for the treatment of iron overload, and 2-hydroxy-1-naphthylaldehyde isonicotinoyl hydrazone (NIH, also known as 311),19,20 an antiproliferative iron scavenger. The biological activity of iron chelators,21 including naturally occurring siderophores such as deferoxamine (DFO, Chart 1, vide infra), is relevant to the study of cancer biology because these compounds interfere with intracellular iron availability and exploit the iron avidity of cancer cells, which require higher levels of this ion to maintain fast proliferation rates.22,23 The involvement of iron chelation in the intracellular chemistry of sirtinol could therefore complicate its biological profile. Herein, we illustrate the iron binding properties of sirtinol as well as its coordination mode. In addition, we investigate whether intracellular iron coordination is a hitherto unrecognized component of the action profile of this sirtuin inhibitor.
Sirtinol was prepared according to published methods24 and isolated as a bright yellow solid. Of the two enantiomers, which present similar inhibitory activity,24 (R)-sirtinol was selected for this investigation. UV-visible absorption changes in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of methanol and buffered water (50 mM PIPES, pH 7.4) indicate that sirtinol coordinates 1 equiv. of divalent Fe(II), the prevalent species in the biological labile iron pool, over a period of 20 min (Fig. S1, ESI†). In order to avoid complications arising from the hydrolytic decomposition of sirtinol in neutral aqueous solutions (as noted by several commercial suppliers of this inhibitor), the stepwise formation of the resulting complex was monitored in dry methanol. Under an inert atmosphere, titration with Fe(II) is associated with spectral changes (Fig. 1) exhibiting approximately isosbestic behavior and saturation at 1 Fe(II) equiv. (Fig. S2, ESI†), thus indicating high affinity and formation of a single species.
:1 mixtures of methanol and buffered water (50 mM PIPES, pH 7.4) indicate that sirtinol coordinates 1 equiv. of divalent Fe(II), the prevalent species in the biological labile iron pool, over a period of 20 min (Fig. S1, ESI†). In order to avoid complications arising from the hydrolytic decomposition of sirtinol in neutral aqueous solutions (as noted by several commercial suppliers of this inhibitor), the stepwise formation of the resulting complex was monitored in dry methanol. Under an inert atmosphere, titration with Fe(II) is associated with spectral changes (Fig. 1) exhibiting approximately isosbestic behavior and saturation at 1 Fe(II) equiv. (Fig. S2, ESI†), thus indicating high affinity and formation of a single species.
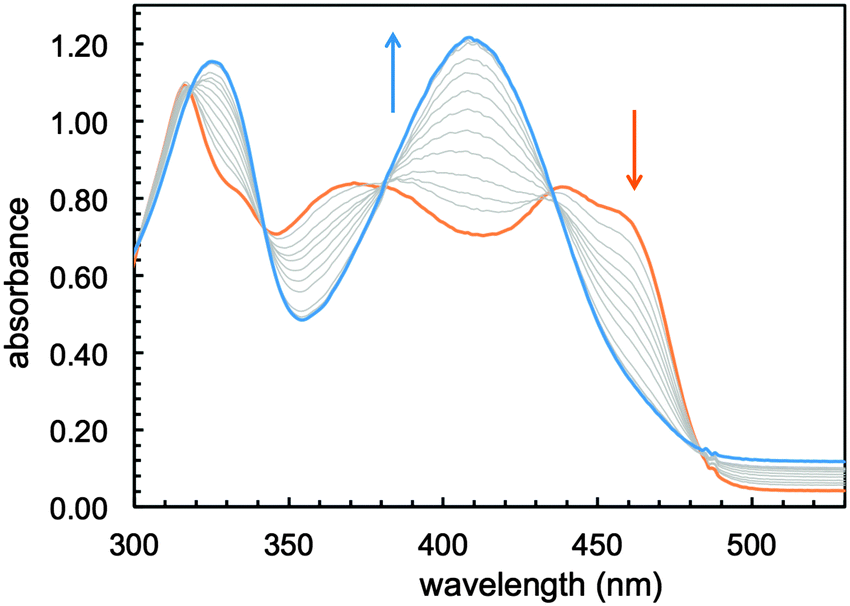 | ||
| Fig. 1 Optical spectral changes associated with the addition of Fe(II) (0.1–1.2 equiv.) to a solution of sirtinol (86 μM, orange trace) in MeOH under an argon atmosphere. | ||
Exposure to air of a stirring solution of the putative Fe(II) sirtinol complex led (over a period of 12 minutes) to formation of a stable green species (Fig. 2) with spectral features analogous to those of the complex obtained in aqueous mixtures (Fig. S1, ESI†). A quantitative determination of thermodynamic iron affinity constants under simulated physiological conditions should take into account the protonation equilibria of the free ligand and its potential complexes of various binding stoichiometries, as well as the redox chemistry of the Fe(II)/Fe(III) couple. In the case of sirtinol, such an investigation would be likely hampered by the hydrolytic decomposition and poor solubility of this compound in water. Nevertheless, this study showed that sirtinol coordinates Fe(II) with high affinity and 1:1 stoichiometry (as indicated by the saturation at 1 equiv.) and that a ferric complex is stabilized under aerobic conditions.
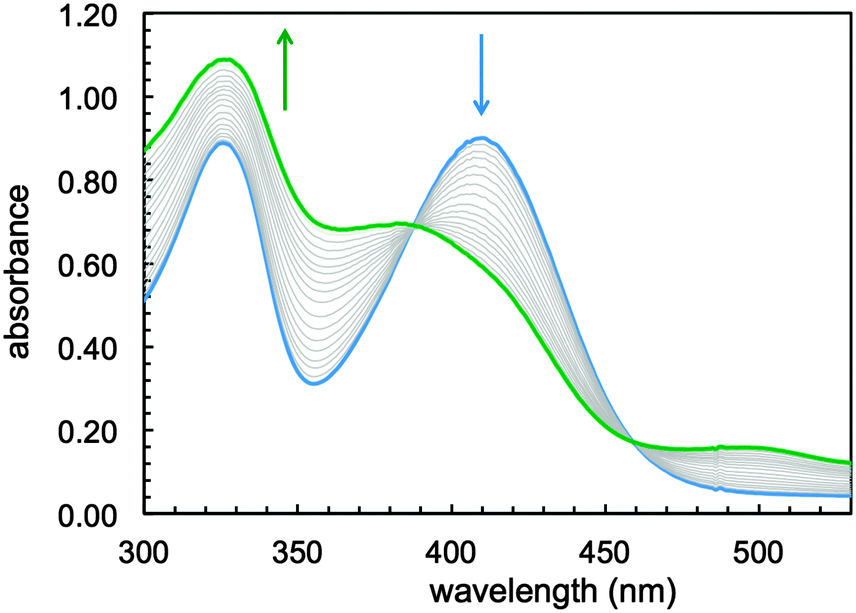 | ||
| Fig. 2 Spectral changes occurring upon air exposure of ferrous sirtinol species ([total sirtinol] = 76 μM, blue trace) leading to oxidation and formation of a ferric species (green trace) in MeOH over a period of 12 min. | ||
The green ferric complex of sirtinol could also be prepared by direct addition of Fe(NO3)3·9H2O to a sirtinol solution (see UV-visible absorption spectrum in Fig. S3, ESI†). [FeIII(sirtinol-H)(NO3)2] was characterized by high-resolution mass spectrometry, elemental analysis, and X-ray diffraction analysis (see ESI† for experimental details). The crystal structure (Fig. 3) revealed the tridentate coordination mode expected for sirtinol, which behaves as a monoanionic ligand. The coordination sphere of the iron center is completed by two nitrato-κ2O ligands in the solid state. The complex has an effective magnetic moment at room temperature of (6.0 ± 0.1)μB, indicating a high-spin configuration for the d5 Fe(III) center. Indeed, the EPR spectrum of this complex is characteristic of a high-spin (S = 5/2) ferric center in rhombic crystal field and shows a major feature at g = 4.3 and a minor feature at g ≈ 9 corresponding to the intermediate and lowest Kramers doublets, respectively (Fig. S4, ESI†).
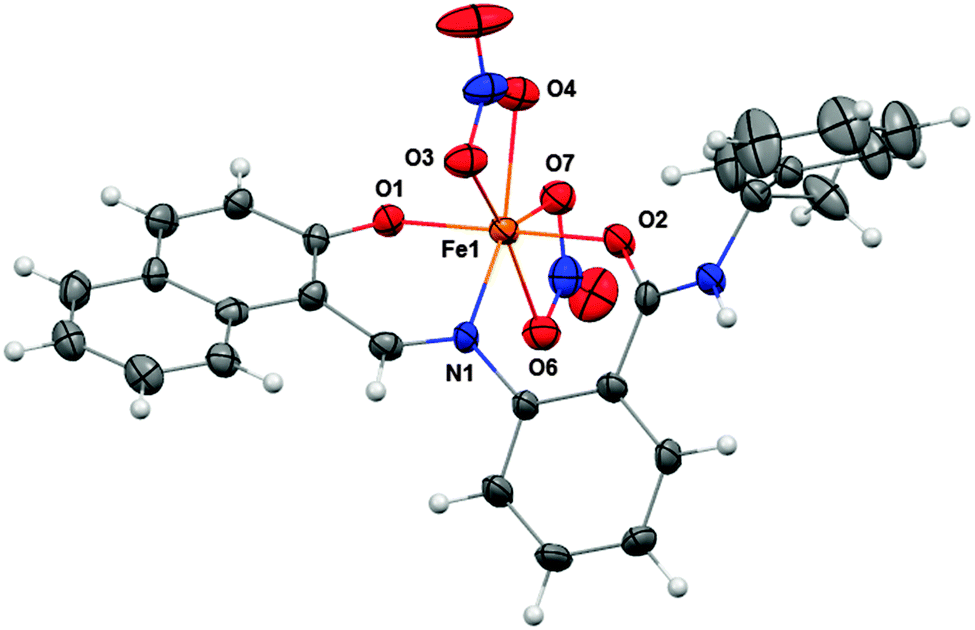 | ||
| Fig. 3 Crystal structure of FeIII(sirtinol-H)(NO3)2 showing a partial labeling scheme. Selected bond lengths (Å): Fe1–O1, 1.889(2); Fe1–O2, 2.011(2); Fe1–N1, 2.072(3). Thermal ellipsoids are scaled to the 50% probability level. CCDC 1036424. | ||
Our chemical characterization data indicate that sirtinol coordinates both Fe(II) and Fe(III) and that a high-spin ferric species is stabilized in solution under aerobic conditions. These findings on the coordination mode and electronic structure of the iron center are in agreement with those previously reported for the structurally similar 2-hydroxynaphthaldehyde derivative NIH (Chart 1), although the latter crystallizes in a 2:1 ligand-to-metal stoichiometry.19‡
As a protein inhibitor and metal chelator, sirtinol bears several features of molecules described as “chemical con artists” because of non-specific abilities (e.g., metal coordination, redox cycling, covalent reactivity) that enhance their profile in screening assays of protein interaction.25 The overall biological activity of these compounds is not simply the result of a drug-like interaction with a specific protein target, but rather a composite of several actions, both independent and/or correlated, in multiple intracellular locales. Inevitably, these promiscuous molecules complicate biological data analysis, and the evaluation of their effective therapeutic potential is often arduous.26 For instance, the multifaceted biological profile of resveratrol, a polyphenol found in red wine and a sirtuin activator, has motivated intense debate over sirtuin proteins in recent years.27–30 In the case of sirtinol, we sought to determine whether iron binding is a component of its intracellular behaviour (in addition to its established function as a sirtuin inhibitor).
The intracellular formation of iron complexes of sirtinol was investigated by EPR spectroscopy in intact leukemia cells following exposure to sirtinol in the growth media. In the g ≈ 2 region of the EPR spectrum (Fig. 4a) of intact frozen cells, the well-studied signal characteristic of the tyrosyl radical within the β2 subunit of active ribonucleotide reductase (RNR) provides an iron-responsive spectroscopic signature. The intensity of this signal, which correlates to enzyme activity, is indeed reduced upon exposure to iron scavengers,31–33 including NIH.34 In our experimental conditions, incubation of Jurkat cells in the presence of siderophore DFO (50 μM, 3 h) or sirtinol (50 μM, 3 h) caused a significant reduction of the amplitude of the Tyr˙ signal compared to that observed for untreated cells (Fig. 4a). On the contrary, exposure to ferric complex [FeIII(sirtinol-H)(NO3)2] (50 μM, 3 h) did not perturb the RNR active site (Fig. 4a, trace 4), indicating that this species does not engage in reactivity affecting the Tyr˙ radical (either directly or through the generation of redox-active species).32
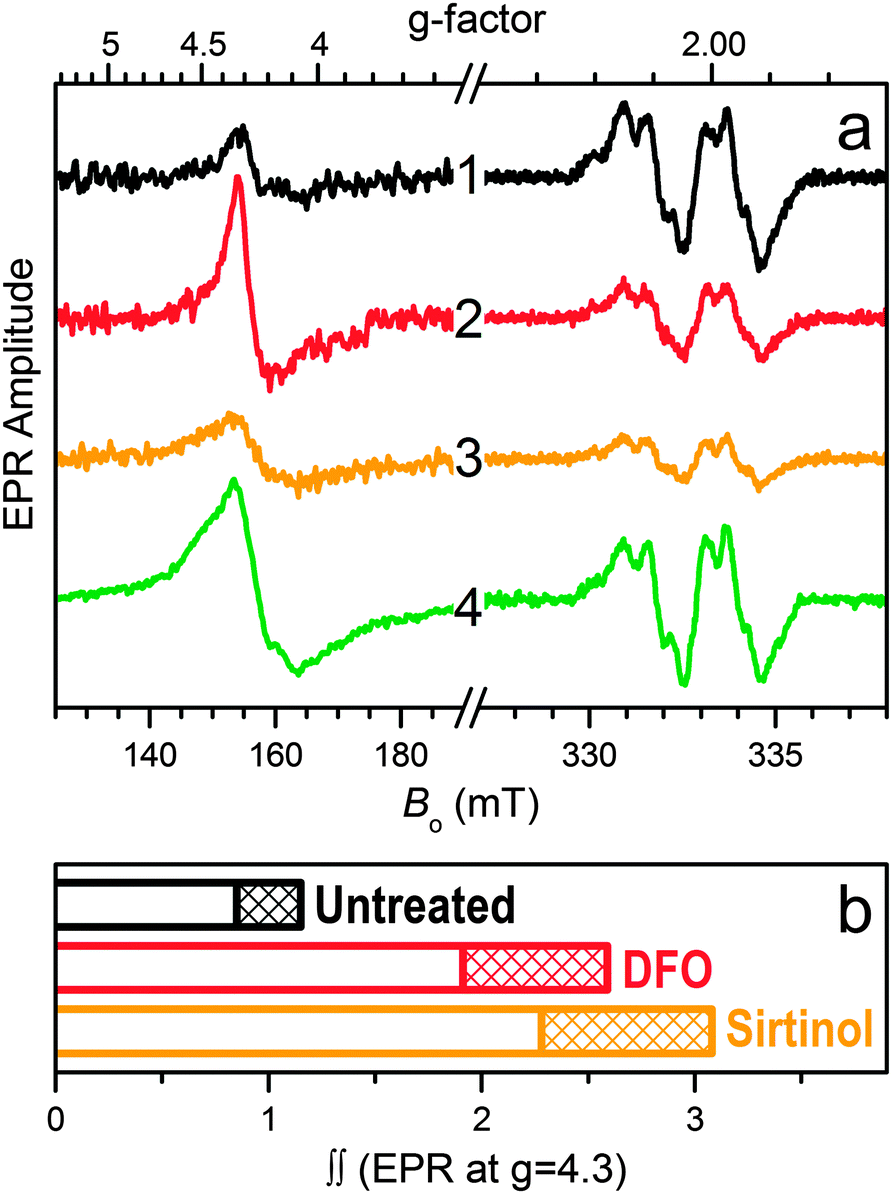 | ||
| Fig. 4 Panel a, EPR spectra of intact Jurkat cells (g = 4.3 and g = 2 regions). Traces 1 through 4 are obtained, respectively, for the untreated sample and for samples treated (50 μM × 3 h) with DFO (2), sirtinol (3), and complex FeIII(sirtinol-H)(NO3)2 (4). Experimental conditions: microwave frequency, 9.337 GHz; microwave power, 2 mW; magnetic field modulation amplitude, 0.5 mT; temperatures, 10 K for g = 4.3 region and 30 K for g = 2 region. Trace 4 in the g = 4.3 region is scaled down by a factor of 1/3. Panel b, relative double integrals of the g = 4.3 signals shown by traces 1–3 in panel a (the double integral value for the untreated cells is set as 1). The patterned regions correspond to the error of ±15% typical for such estimates. For comparison, the double integral value for g = 4.3 region of unscaled trace 4 (not shown) is 20 ± 3. | ||
Because sirtinol is similar to DFO in its stabilization of ferric centers upon iron coordination, both chelators were expected to increase the intracellular amount of high-spin Fe(III) centers. Indeed, the EPR data for these whole-cell samples in the g = 4.3 region (Fig. 4a) confirmed that exposure to sirtinol (or DFO) leads to formation of ferric species in cultured cells. The relative double integrals of these signals (which are proportional to the relative concentrations of the corresponding ferric centers) for the untreated sample and those treated with the chelators (Fig. 4b) demonstrate the increase in the intracellular high-spin ferric species upon treatment with DFO (as previously reported)31 and sirtinol. Although the amplitude of the EPR signal at g ∼ 4.3 for the intracellular sirtinol complex (trace 3 in Fig. 4a) is smaller than that of the complex with DFO (trace 2), the double integral of the sirtinol signal is greater (Fig. 4b). This effect is explained by a greater EPR linewidth of the sirtinol complex (caused by departures of the crystal field rhombicity, E/D, from 1/3), which is much easier to observe for the spectrum of synthetic FeIII(sirtinol-H)(NO3)2 (trace 4 in Fig. 4a) because of its significantly larger amplitude.
Our findings demonstrate the iron binding ability of sirtinol, which behaves as a high-affinity tridentate chelator in organic solvents and stabilizes a ferric center under aerobic conditions. In cultured cells, sirtinol affects highly regulated iron levels through formation of ferric complexes. Similarly to known antiproliferative chelator and RNR inhibitor DFO,31 sirtinol leads to a reduction in the EPR signal associated to the Tyr˙ residue in RNR, a radical species that is essential for enzymatic activity in DNA biosynthesis. The ability of sirtinol to bind other intracellular metal cations (e.g., copper, zinc) and the effect of metal binding on the inhibitory activity with respect to sirtuins remain to be determined. Nevertheless, intracellular iron chelation affects cell growth and proliferation,35 and could thus influence overall readouts in cell-based assays involving sirtinol. In line with the inclusion of several metal-binding compounds among the pan-assay interference compounds (PAINS)25,26 in screening assays, our findings underscore the importance of metal interactions in the discovery of protein inhibitors and activators. A full understanding of these effects is not only critical for the definition of clean activity profiles, but also desirable for the potential engineering of polypharmacologic networks and of protein–small-molecule interactions that are modulated dynamically by metal coordination.
This work was supported by the University of Arizona. We thank Dr Sue Roberts for assistance with the acquisition and analysis of X-ray diffraction data. A.V.A. gratefully acknowledges the NSF (grants DBI-0139459, DBI-9604939, BIR-9224431) and NIH (grants S10RR020959, S10RR026416-01) for the development of the EPR facility at the University of Arizona.
Notes and references
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Chem. Educ., 2013, 90, 1403 CrossRef CAS.
- K. J. Franz, Curr. Opin. Chem. Biol., 2013, 17, 143 CrossRef CAS PubMed.
- R. H. Houtkooper, E. Pirinen and J. Auwerx, Nat. Rev. Mol. Cell Biol., 2012, 13, 225 CAS.
- M. Roth and W. Y. Chen, Oncogene, 2014, 33, 1609 CrossRef CAS PubMed.
- B. J. Morris, Free Radical Biol. Med., 2013, 56, 133 CrossRef CAS PubMed.
- D. A. Sinclair and L. Guarente, Annu. Rev. Pharmacol. Toxicol., 2014, 54, 363 CrossRef CAS PubMed.
- L. Chen, Curr. Med. Chem., 2011, 18, 1936 CrossRef CAS.
- H. Yuan and R. Marmorstein, J. Biol. Chem., 2012, 287, 42428 CrossRef CAS PubMed.
- C. M. Grozinger, E. D. Chao, H. E. Blackwell, D. Moazed and S. L. Schreiber, J. Biol. Chem., 2001, 276, 38837 CrossRef CAS PubMed.
- H. Ota, E. Tokunaga, K. Chang, M. Hikasa, K. Iijima, M. Eto, K. Kozaki, M. Akishita, Y. Ouchi and M. Kaneki, Oncogene, 2006, 25, 176 CAS.
- B. Peck, C.-Y. Chen, K.-K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C.-D. Hsiao and E. W. F. Lam, Mol. Cancer Ther., 2010, 9, 844 CrossRef CAS PubMed.
- A. M. Kalle, A. Mallika, J. Badiger, Alinakhi, P. Talukdar and Sachchidanand, Biochem. Biophys. Res. Commun., 2010, 401, 13 CrossRef CAS PubMed.
- D. Rotili, D. Tarantino, A. Nebbioso, C. Paolini, C. Huidobro, E. Lara, P. Mellini, A. Lenoci, R. Pezzi, G. Botta, M. Lahtela-Kakkonen, A. Poso, C. Steinkuehler, P. Gallinari, R. De Maria, M. Fraga, M. Esteller, L. Altucci and A. Mai, J. Med. Chem., 2012, 55, 10937 CrossRef CAS PubMed.
- J. Wang, T. H. Kim, M. Y. Ahn, J. Lee, J. H. Jung, W. S. Choi, B. M. Lee, K. S. Yoon, S. Yoon and H. S. Kim, Int. J. Oncol., 2012, 41, 1101 CAS.
- T. Kozako, A. Aikawa, T. Shoji, T. Fujimoto, M. Yoshimitsu, S. Shirasawa, H. Tanaka, S.-i. Honda, H. Shimeno, N. Arima and S. Soeda, Int. J. Cancer, 2012, 131, 2044 CrossRef CAS PubMed.
- B.-J. Hwang, A. Madabushi, J. Jin, S.-Y. S. Lin and A. L. Lu, Am. J. Cancer Res., 2014, 4, 211 Search PubMed.
- T. T. Y. Wang, N. W. Schoene, E.-K. Kim and Y. S. Kim, Mol. Carcinog., 2013, 52, 676 CrossRef CAS PubMed.
- G. Shashaty, R. Frankewich, T. Chakraborti, J. Choudary, S. Al-Fayoumi, A. Kacuba, S. Castillo, K. Robie-Suh, D. Rieves, K. Weiss and R. Pazdur, Oncology, 2006, 20, 1799 Search PubMed.
- D. R. Richardson and P. V. Bernhardt, JBIC, J. Biol. Inorg. Chem., 1999, 4, 266 CrossRef CAS.
- T. B. Chaston, D. B. Lovejoy, R. N. Watts and D. R. Richardson, Clin. Cancer Res., 2003, 9, 402 CAS.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547 CrossRef CAS PubMed.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342 CrossRef CAS PubMed.
- D. R. Richardson, D. S. Kalinowski, S. Lau, P. J. Jansson and D. B. Lovejoy, Biochim. Biophys. Acta, 2009, 1790, 702 CrossRef CAS PubMed.
- A. Mai, S. Massa, S. Lavu, R. Pezzi, S. Simeoni, R. Ragno, F. R. Mariotti, F. Chiani, G. Camilloni and D. A. Sinclair, J. Med. Chem., 2005, 48, 7789 CrossRef CAS PubMed.
- J. Baell and M. A. Walters, Nature, 2014, 513, 481 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719 CrossRef CAS PubMed.
- B. P. Hubbard, A. P. Gomes, H. Dai, J. Li, A. W. Case, T. Considine, T. V. Riera, J. E. Lee, E. S. Yen, D. W. Lamming, B. L. Pentelute, E. R. Schuman, L. A. Stevens, A. J. Y. Ling, S. M. Armour, S. Michan, H. Z. Zhao, Y. Jiang, S. M. Sweitzer, C. A. Blum, J. S. Disch, P. Y. Ng, K. T. Howitz, A. P. Rolo, Y. Hamuro, J. Moss, R. B. Perni, J. L. Ellis, G. P. Vlasuk and D. A. Sinclair, Science, 2013, 339, 1216 CrossRef CAS PubMed.
- S. J. Park, F. Ahmad, A. Philp, K. Baar, T. Williams, H. B. Luo, H. M. Ke, H. Rehmann, R. Taussig, A. L. Brown, M. K. Kim, M. A. Beaven, A. B. Burgin, V. Manganiello and J. H. Chung, Cell, 2012, 148, 421 CrossRef CAS PubMed.
- M. Pacholec, J. E. Bleasdale, B. Chrunyk, D. Cunningham, D. Flynn, R. S. Garofalo, D. Griffith, M. Griffor, P. Loulakis, B. Pabst, X. Y. Qiu, B. Stockman, V. Thanabal, A. Varghese, J. Ward, J. Withka and K. Ahn, J. Biol. Chem., 2010, 285, 8340 CrossRef CAS PubMed.
- J. A. Baur, K. J. Pearson, N. L. Price, H. A. Jamieson, C. Lerin, A. Kalra, V. V. Prabhu, J. S. Allard, G. Lopez-Lluch, K. Lewis, P. J. Pistell, S. Poosala, K. G. Becker, O. Boss, D. Gwinn, M. Y. Wang, S. Ramaswamy, K. W. Fishbein, R. G. Spencer, E. G. Lakatta, D. Le Couteur, R. J. Shaw, P. Navas, P. Puigserver, D. K. Ingram, R. de Cabo and D. A. Sinclair, Nature, 2006, 444, 337 CrossRef CAS PubMed.
- C. E. Cooper, G. R. Lynagh, K. P. Hoyes, R. C. Hider, R. Cammack and J. B. Porter, J. Biol. Chem., 1996, 271, 20291 CrossRef CAS PubMed.
- Y. Aye, M. J. C. Long and J. Stubbe, J. Biol. Chem., 2012, 287, 35768 CrossRef CAS PubMed.
- E. A. Akam, T. M. Chang, A. V. Astashkin and E. Tomat, Metallomics, 2014, 6, 1905 RSC.
- D. A. Green, W. E. Antholine, S. J. Wong, D. R. Richardson and C. R. Chitambar, Clin. Cancer Res., 2001, 7, 3574 CAS.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, X-ray diffraction analysis, UV-visible absorption and EPR spectroscopic data, cell culture details and whole-cell EPR sample preparation. CCDC 1036424. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00829h |
| ‡ The formation of a sirtinol complex of 2:1 binding stoichiometry was investigated in organic solvents upon addition of 0.5 equiv. Fe(III). Although the formation of both 2:1 and 1:1 complexes was supported by mass spectrometry data, multiple crystallization attempts ultimately led to the isolation of the 1:1 complex shown in Fig. 3. |
| This journal is © The Royal Society of Chemistry 2015 |