
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of a 1,4-diketone intermediate in oxidative dimerization of 2-hydroxyanthracene and its conversion to oxahelicene†
Takashi
Matsuno
a,
Yutaro
Koyama
a,
Satoru
Hiroto
*a,
Jatish
Kumar
b,
Tsuyoshi
Kawai
b and
Hiroshi
Shinokubo
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: hshino@apchem.nagoya-u.ac.jp; hiroto@apchem.nagoya-u.ac.jp
bGraduate School of Material Science, Nara Institute of Science and Technology (NAIST), Ikoma, 630-0192, Japan
First published on 9th February 2015
Abstract
Oxidation of 2-hydroxy-9,10-dialkynylanthracenes resulted in highly regioselective dimerization to furnish metastable dearomatized 1,4-diketones rather than stable aromatic diols. The 1,4-diketones were converted to oxahelicenes, which exhibited strong fluorescence both in solution and solid state as well as chiroptical properties.
2,2′-Dihydroxy-1,1′-binaphthyl (BINOL) is one of the most versatile chiral scaffolds in asymmetric synthesis, which is frequently employed to create a variety of metal and non-metal chiral catalysts. Oxidative coupling of 2-naphthol and other derivatives has been a useful methodology to provide BINOL and other axially chiral aromatic diols. The mechanism of oxidative coupling has been considered in the following three processes: (i) homolytic coupling of two radicals (A˙ + A˙), (ii) heterolytic coupling (A+ + A) and (iii) radical–anion coupling (A˙ + A−).1–3 In the radical–radical coupling process, formation of a diketone intermediate is expected through dimerization of a carbon-centred radical contributor of the 2-naphthoxyl radical (Scheme 1). However, such a dearomatized 1,4-diketone has been an elusive intermediate due to its instability. The 1,4-diketone rapidly undergoes tautomerization to the more stable enol form to yield BINOL.
 | ||
| Scheme 1 Plausible reaction mechanism of BINOL formation via the radical–radical coupling process. | ||
During the course of our study on oxidation of aromatic alcohols,4 we unexpectedly isolated such metastable 1,4-diketone intermediates 2 by oxidation of 2-hydroxyanthracenes 1. Although oxidative dimerization of 2-hydroxyanthracenes to BINOL-like axially chiral diols has been reported, dearomatized 1,4-diketone species have never been isolated.5 We have also demonstrated the synthesis of oxahelicenes 6 from 1,4-diketones 2, which exhibit bright fluorescence both in solution and solid state as well as chiroptical properties.
We found that oxidation of 2-hydroxy-9,10-bis(triethylsilylethynyl)anthracene 1a with MnO2 in dichloromethane at room temperature afforded dimeric diketone 2a in 63% yield without formation of diol 4 (Scheme 2). The structure of 2a was assigned on the basis of NMR and MS analyses. The 1H NMR spectrum of 1a exhibited olefinic signals at 8.17 and 6.35 ppm as well as a methine proton signal at 4.35 ppm. The presence of carbonyl groups was confirmed by the peak at 200.9 ppm, as shown in its 13C NMR spectrum. Triisopropylsilyl (TIPS) substituted diketone 2b was similarly prepared in 50% yield. In this case, ortho-quinone 3b was obtained as a byproduct. The dimerization also proceeded with less bulky tert-butyl substituent 1c to provide 2c in 84% yield. The improvement of the yield is probably due to steric reason. Diketones 2 were relatively stable but treatment with KOtBu resulted in tautomerization to afford the more stable enol-form 4 in 59% yield (Scheme 2).
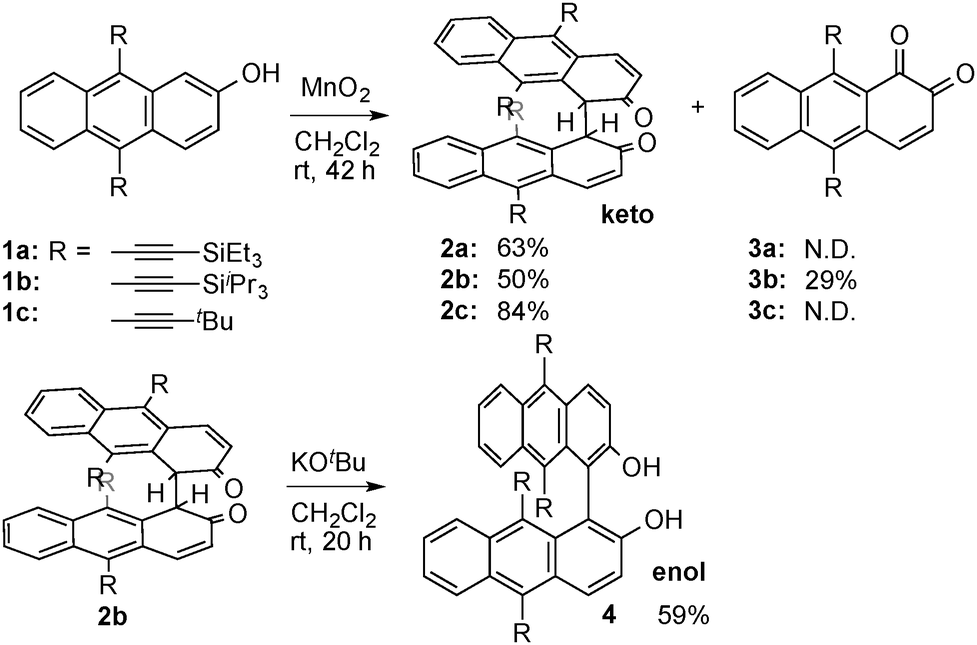 | ||
| Scheme 2 Oxidation of 2-hydroxyanthracenes 1. | ||
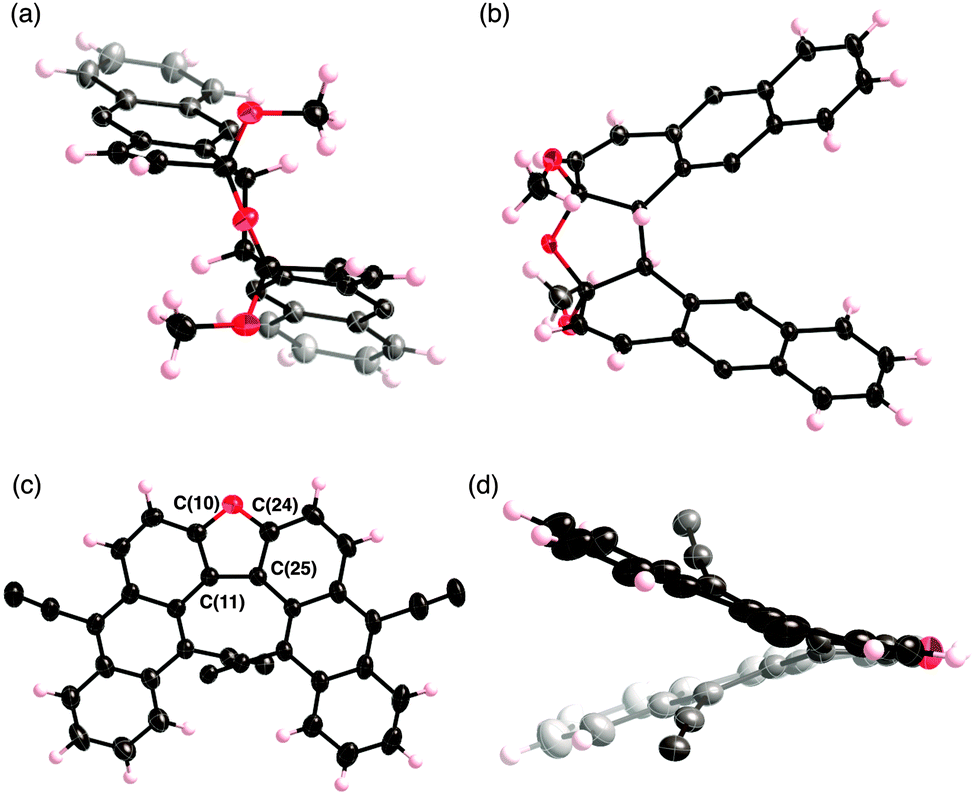
The structures of 2b and 4 were unambiguously elucidated by X-ray diffraction analysis (Fig. 1). In the case of 2b, two anthracene rings overlap in a face-to-face fashion. Notably, the bond length of the bridging carbon–carbon linkage is 1.62 Å, which is considerably longer than that of the standard Csp3–Csp3 bond (1.54 Å). This elongation is caused by columbic repulsion between two anthracene rings. This could be a reason for facile bond cleavage of 2b to 1b during silica-gel column chromatography. In the case of 4, two anthracene units cross each other at an angle of 102.7°, which is larger than that of BINOL.6 The alkynyl groups are substantially bent due to the steric repulsion with anthracene moieties.
 | ||
| Fig. 1 X-ray crystal structures of 2b and 4. (a) Top view and (b) side view of 2b and (c) side view and (d) front view of 4. The ethynyl groups are omitted in (a) and (b), and iPr groups are omitted in (c) and (d) for clarity. The thermal ellipsoids are scaled at the 50% probability level. | ||
The isolation of the keto-form 2 was intriguing because BINOL derivatives usually exhibit no keto–enol tautomerism due to the instability of keto-forms.7 The distorted structure of 4 should be a clue for the relative stabilization of the keto-form 2 because no keto–enol tautomerization could be observed for the corresponding monomer 1. The large displacement of conformations between 2b and 4 should increase the conversion barrier from 2b to 4, which is one of the key factors for the stability of 2b. Moreover, the calculated LUMO of 2b spread over the entire molecule, indicating effective delocalization of π-electrons (Fig. S32, ESI†). This is also suggested by optical analysis. In comparison to 1,1-dimethoxy-2-oxoanthracene, a slight but obvious red-shift of the lowest energy band was observed for 2b (Fig. S28, ESI†), indicating the presence of electronic communication between two anthracene units in 2b. This interaction between anthracenes would also contribute to the stabilization of the keto-form 2b.
We then attempted construction of the fused furan ring to obtain oxahelicenes 6.8 Helical-shaped π-conjugated molecules have attracted tremendous interest as promising candidates for new-age functional materials.9,10 Their rigid and twisted π-condensed ring systems induce chiroptical properties such as enhanced CD signals and circularly polarized luminescence (CPL).11 They, however, commonly have low emission efficiency due to their ortho-condensed structures, which result in rapid intersystem crossing from an excited singlet state to a triplet state.12
We found that treatment of 2a with TsOH·H2O and methyl orthoformate in methanol yielded 57% of diacetal 5a. Methoxy groups were readily eliminated by treatment with CF3SO3H to furnish oxahelicene 6a in 16% yield (Scheme 3). In the case of TIPS derivative 2b, the yield of 6b was improved to 47% due to the higher stability of the alkyne group under acidic conditions. Unfortunately, the corresponding cyclic diacetal 5 was not obtained from 2c.
 | ||
| Scheme 3 Conversion of 2 to oxahelicenes 6. | ||
The structures of 5b and 6b were confirmed by X-ray diffraction analysis (Fig. 2). The X-ray crystal structure revealed a distorted helical conformation of 6b. As shown, two anthracene units and two ethynyl groups are bent due to severe steric repulsion. The dihedral angle between the two naphthalene rings is 40.9°.
 | ||
| Fig. 2 X-ray crystal structures of 5b and 6b: (a) top view and (b) side view of 5b; (c) top view and (d) side view of 6b. The silylethynyl groups in (a) and (b) and triisopropylsilyl groups in (c) and (d) are omitted for clarity. The thermal ellipsoids are scaled at the 50% probability level. | ||
Fig. 3 displays UV/vis absorption spectra of 1b, 4, 6a and 6b and fluorescence spectra of 6a and 6b in dichloromethane solutions. The dimer 4 exhibited a bathochromic shift compared to 1b as commonly seen in perpendicularly linked acene dimers.13 A larger shift was observed for 6b, indicating effective π-conjugation over two anthracene units. No significant difference between 6a and 6b was observed. On the other hand, emission spectra exhibited clear differences. The emission of 6a appeared at 558 nm with Φf = 0.45. Slight blue-shifted and enhanced emission (Φf = 0.66) was observed for 6b. This difference can be accounted for by the higher flexibility of 6a, enhancing non-radiative decay, judging from the larger Stokes shift of 6a (Δν = 1803 cm−1). These results indicate that the substituent can control the emission feature of helicene derivatives.
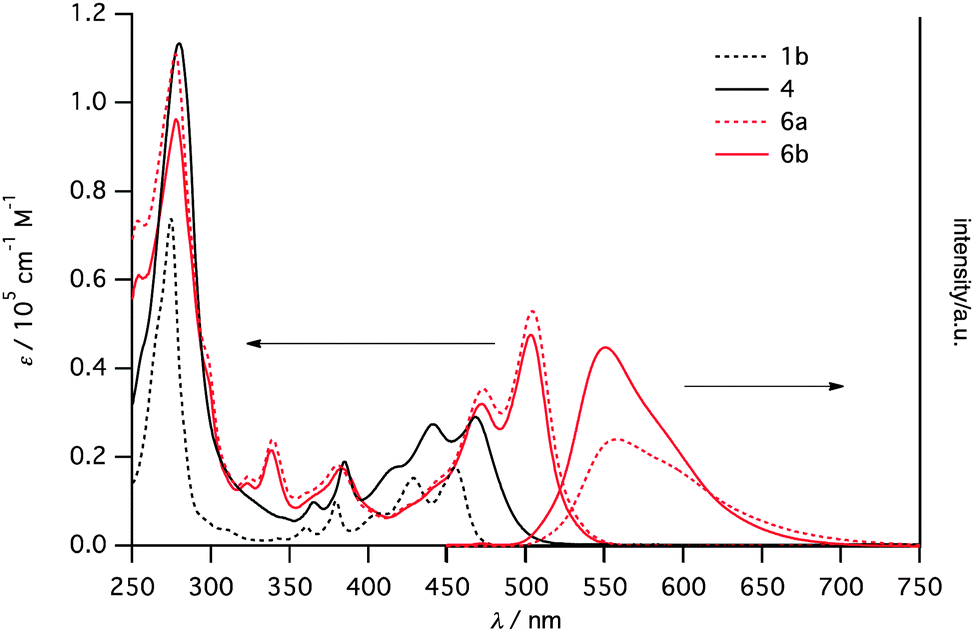 | ||
| Fig. 3 UV/vis absorption spectra of 1b (black, dotted), 4 (black, solid) 6a (red dotted) and 6b (red solid), and emission spectra of 6a (red, dotted) and 6b (red, solid). All spectra were recorded in dichloromethane. | ||
The chiroptical properties of 6b were also investigated. Optical resolution was accomplished by HPLC using a CHIRALPAK® IF column. Both enantiomers were stable enough and underwent no racemization at room temperature. The CD spectrum of the first eluted fraction exhibited a (+) Cotton effect and that of the other isomer exhibited a (−) Cotton effect as the mirror image (Fig. 4). The absolute structure of the latter enantiomer was proven to be a (P)-helix on the basis of X-ray diffraction analysis (Fig. S30, ESI†) and TD-DFT calculations. Furthermore, both enantiomers exhibited CPL with glum = 1.2 × 10−3. This value is consistent with heterohelicenes and relatively high for small organic molecules.
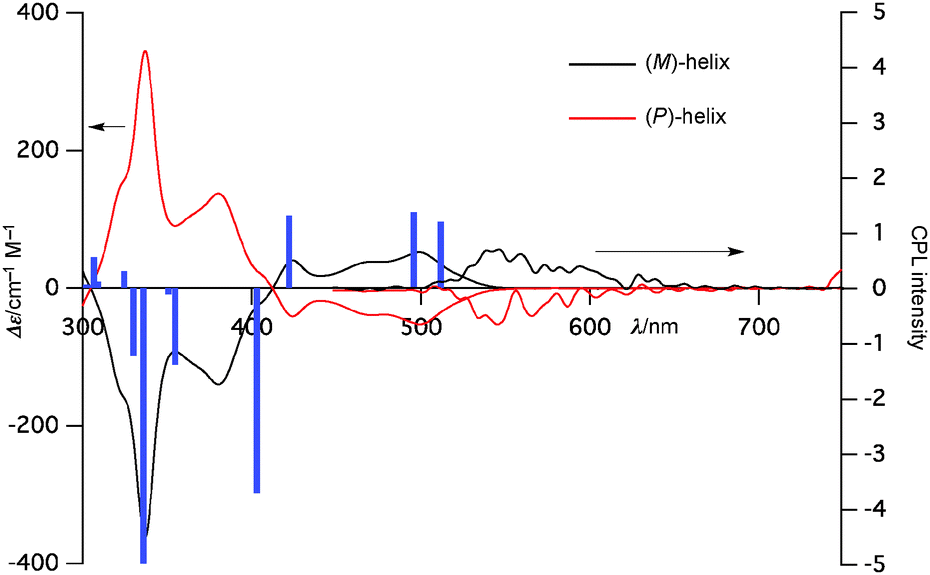 | ||
| Fig. 4 CD and CPL spectra of 6b. Blue bars represent oscillator strengths for the (M)-helix enantiomer estimated by the TD-DFT method. All spectra were recorded in dichloromethane solution (c = 1.0 × 10−5 M). | ||
Interestingly, oxahelicene 6b exhibited intense emission in the solid state (Fig. 5). A racemic microcrystalline sample of 6b showed strong fluorescence (Φf = 0.41). This fluorescence quantum yield in the solid state is the highest among helicene derivatives reported previously.14 The emission was reduced for enantiomerically pure crystals of 6-(P) (Φf = 0.14). Reduction in the quantum yield was due to differences in the crystal packing between racemic 6b and its (P)-isomer 6-(P), suggested by the red-shifted spectrum of 6-(P) (λem = 581 nm) as compared with 6b (λem = 574 nm). In the crystal packing of 6b, a pair of (P)- and (M)-enantiomers are placed in a face-to-face fashion to form a coplanar dimer (Fig. S31, ESI†). In contrast, molecules in 6-(P) are aligned in a herringbone fashion.
 | ||
| Fig. 5 Emission spectra of a racemic mixture (red) and a (P)-enantiomer (blue) of 6b in the solid state (microcrystals). | ||
In summary, we have discovered that oxidation of 2-hydroxyanthracenes furnished dearomatized diketones 2 as metastable intermediates. Treatment of 2b with a base or heating induced tautomerization into bianthranyl diol 4. We have also achieved synthesis of oxahelicenes 6 from 2 in two steps under mild conditions. Oxahelicene 6b exhibited strong emission and CPL activity. This work demonstrates that oxidation of hydroxylated oligoacenes offers an easy access to distorted π-conjugated molecules with fascinating structures and optical characteristics.
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas “Science of Atomic Layers” (26107519) and “pi-System Figuration” (26102003) and Program for Leading Graduate Schools “Integrative Graduate Education and Research in Green Natural Sciences”, MEXT, Japan. H.S. acknowledges the financial support from Yamada Science Foundation.
Notes and references
- (a) Q.-X. Guo, Z.-J. Wu, Z.-B. Luo, Q.-Z. Liu, J.-L. Ye, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Am. Chem. Soc., 2007, 129, 13927 CrossRef CAS PubMed; (b) M. Matsushita, K. Kamata, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 6632 CrossRef CAS PubMed; (c) X. Li, J. B. Hewgley, C. A. Mulrooney, J. Yang and M. C. Kozlowski, J. Org. Chem., 2003, 68, 5500 CrossRef CAS PubMed.
- M. Hovorka and J. Závada, Tetrahedron, 1992, 48, 9517 CrossRef CAS.
- (a) H. Egami, K. Matsumono, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633 CrossRef CAS PubMed; (b) M. Smrčina, Š. Vyskočil, B. Máca, M. Polásek, T. A. Claxton, A. P. Abbott and P. Kočovský, J. Org. Chem., 1994, 59, 2156 CrossRef.
- Y. Koyama, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2013, 52, 5740 CrossRef CAS PubMed.
- (a) S. Zhang, Y. Wang, Z. Song, K. Nakajima and T. Takahashi, Chem. Lett., 2013, 42, 697 CrossRef CAS; (b) S. Takizawa, J. Kodera, Y. Yoshida, M. Sako, S. Breukers, D. Enders and H. Sasai, Tetrahedron, 2014, 70, 1786 CrossRef CAS PubMed.
- BINOL has a cisoid conformation (dihedral angle < 90°) in crystal. (a) K. Mori, Y. Masuda and S. Kashino, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 1224 CrossRef; (b) L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS PubMed.
- Keto–enol tautomerism of 1,4-naphthalenediols was extensively investigated. (a) E. P. Kündig, A. E. García, T. Lomberget and G. Bernardinelli, Angew. Chem., Int. Ed., 2006, 45, 98 CrossRef PubMed; (b) H. Laatsch, Liebigs Ann. Chem., 1980, 140 CrossRef CAS; (c) M. S. Pearson, B. J. Jensky, F. X. Greer, J. P. Hagstrom and N. M. Wells, J. Org. Chem., 1978, 43, 4617 CrossRef CAS . However, no keto tautomer of monohydroxyarenes has been isolated.
- (a) R. Irie, A. Tanoue, S. Urakawa, T. Imahori, K. Igawa, T. Matsunoto, K. Tomooka, S. Kikuta, T. Uchida and T. Katsuki, Chem. Lett., 2011, 40, 1343 CrossRef CAS; (b) M. Salim, A. Akutsu, T. Kimura, M. Minabe and M. Karikomi, Tetrahedron Lett., 2011, 52, 4518 CrossRef CAS PubMed; (c) J. F. Schneider, M. Nieger, K. Nättinen and K. H. Dötz, Synthesis, 2005, 1109 CAS.
- For reviews, see: (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680 RSC; (b) M. Gingras, Chem. Soc. Rev., 2013, 42, 968 RSC; (c) Z. Peng and N. Takenaka, Chem. Rec., 2013, 13, 28 CrossRef CAS PubMed; (d) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed; (e) R. Amemiya and M. Yamaguchi, Chem. Rec., 2008, 8, 116 CrossRef CAS PubMed; (f) A. Rajca and M. Miyasaka, in Functional Organic Materials: Syntheses, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007, p. 543 Search PubMed; (g) C. Nuckolls, R. Shao, W.-G. Jang, N. A. Clark, D. M. Walba and T. J. Katz, Chem. Mater., 2002, 14, 773 CrossRef CAS; (h) T. J. Katz, Angew. Chem., Int. Ed., 2000, 39, 1921 CrossRef CAS.
- For recent edge-cutting reports on helicene-like molecules, see: (a) D. Schweinfurth, M. Zalibera, M. Kathan, C. Shen, M. Mazzolini, N. Trapp, J. Crassous, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2014, 136, 13045 CrossRef CAS PubMed; (b) Y. Kimura, N. Fukawa, Y. Miyauchi, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 8480 CrossRef CAS PubMed; (c) L. Pospíšil, L. Bednárová, P. Štépánek, P. Slavíček, J. Vávra, M. Hromadvá, H. Dlouhá, J. Tarábek and F. Teplý, J. Am. Chem. Soc., 2014, 136, 10826 CrossRef PubMed; (d) D. Mendola, N. Saleh, N. Vanthuyne, C. Roussel, L. Toupet, F. Castiglione, T. Caronna, A. Mele and J. Crassous, Angew. Chem., Int. Ed., 2014, 53, 5786 CrossRef CAS PubMed; (e) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915 RSC.
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555 CrossRef CAS PubMed and references are therein.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899 CrossRef CAS.
- K. Tanaka, N. Aratani, D. Kuzuhara, S. Sakamoto, T. Okujima, N. Ono, H. Uno and H. Yamada, RSC Adv., 2013, 3, 15310 RSC.
- H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2013, 15, 2104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data for all new compounds, photophysical data and crystallographic data of 2b, 4, 5b, 6b and 6-(P). CCDC 1041270–1041274. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00764j |
| This journal is © The Royal Society of Chemistry 2015 |