
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic CO2 reduction by a mixed metal (Zr/Ti), mixed ligand metal–organic framework under visible light irradiation†
Yeob
Lee
a,
Sangjun
Kim
b,
Jeung Ku
Kang
*bc and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
bDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 305-701, Republic of Korea. E-mail: jeung@kaist.ac.kr
cGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 305-701, Republic of Korea
First published on 18th February 2015
Abstract
Postsynthetic exchange (PSE) of Ti(IV) into a Zr(IV)-based MOF enabled photocatalytic CO2 reduction to HCOOH under visible light irradiation with the aid of BNAH and TEOA. Use of a mixed-ligand strategy enhanced the photocatalytic activity of the MOF by introducing new energy levels in the band structure of the MOF.
Carbon dioxide (CO2) produced by energy generation based on fossil fuels contributes to global warming and consequently negative effects on the environment.1–3 Direct conversion of CO2 into useful chemicals is regarded as a promising technology for addressing the CO2 problem.4 Inspired by nature, systems that can photocatalytically generate hydrocarbon fuels from CO2 have gathered substantial interest. Homogeneous systems based on transition metal centers with photosensitizers can show high efficiencies, but they are generally not reusable.5 On the other hand, heterogeneous photocatalysts based on semiconductors are robust, but suffer from low efficiencies because most of these systems only absorb UV light, which represents only ∼4% of the solar energy spectrum.6 Developing new photocatalysts that can harvest more of the solar spectrum, while retaining high stability and efficiency, would be an important advancement for CO2 utilization.
Metal–organic frameworks (MOFs) are crystalline hybrid materials for CO2 conversion with high specific surface area, because they can be tuned by design.7–10 The use of MOFs in photocatalysis of CO2 has been investigated.11,12 Fu et al. found that Ti-based NH2-MIL-125(Ti) could mediate the conversion of CO2 to HCOO− in the presence of triethanolamine (TEOA). In addition, they found that visible light sensitivity could be introduced by incorporating 2-aminobenzene-1,4-dicarboxylic acid (NH2-bdc).11 Another work also used NH2-MIL-125(Ti) to produce H2 from a TEOA–H2O solution under visible light irradiation.13
Now a well studied, robust, and highly porous MOF, UiO-66 (UiO = University of Oslo) constructed from Zr secondary-building units (SBUs) and benzene-1,4-dicarboxylic acid (H2bdc),14–18 cannot catalyze the reduction of CO2 to HCOO− or the reduction of H2O to H2. The Zr6 SBUs (Zr6O4(OH)4) cannot accept electrons from the bdc linker under light irradiation (unlike the aforementioned Ti8 SBUs (Ti8O8(OH)4)) because the redox potential energy level of the Zr6 SBUs in UiO-66 lies above the LUMO of the bdc ligands.12,13,19 Consequently, it was hypothesized that embedding Ti ions into the Zr6 SBUs of UiO-66 might introduce catalytic activity to UiO-66 by lowering the redox potential energy of the Zr6 cluster.
Herein, we report a mixed-ligand, mixed-metal UiO-66-derivative (Zr4.3Ti1.7O4(OH)4(C8H7O4N)5.17(C8H8O4N2)0.83, 1(Zr/Ti)) obtained by postsynthetic exchange (PSE)20–26 as an effective photocatalyst for CO2 reduction under visible light irradiation. The Ti(IV) ions make the SBUs capable of accepting electrons generated via light absorption by the organic linkers. Introducing a small amount of 2,5-diaminobenzene-1,4-dicarboxylic acid ((NH2)2-bdc) as a co-ligand provided new energy levels in the band structure of the MOF and introduced broader light absorption coverage for the MOF (Scheme 1).
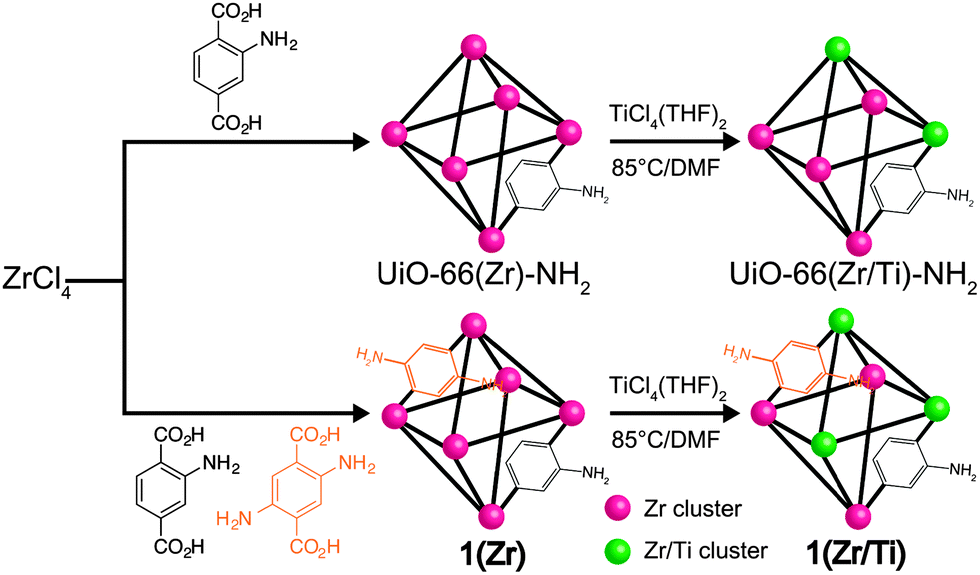 | ||
| Scheme 1 Synthesis of mixed-ligand MOF 1(Zr)via PSE to obtain mixed-metal MOFs 1(Zr/Ti), UiO-66(Zr/Ti)-NH2. | ||
Solvothermal synthesis of 1(Zr) produced nanocrystals with excellent crystallinity (Fig. 1) and a narrow size distribution (Fig. S1, ESI†). The crystal growth of 1(Zr) gave particles with an edge length of ∼70 nm, which is smaller than that obtained for UiO-66(Zr)-NH2 at around ∼200 nm. 1(Zr) absorbed visible light as shown by two broad absorption bands in the UV-vis spectrum, while UiO-66(Zr)-NH2 absorbs only a small portion of blue light beyond the UV spectrum (Fig. S2, ESI†). The ratio between NH2-bdc and (NH2)2-bdc was calculated by dissolving the MOF under alkaline conditions and measuring the 1H NMR spectrum in solution (see ESI†). 1(Zr) contained ∼14% (NH2)2-bdc and 86% NH2-bdc, which is slightly different from the ligand ratio used in synthesis of the MOF (this may be due to the lower solubility of (NH2)2-bdc in N,N-dimethylformamide (DMF)). Afterwards, these Zr-based MOFs were exposed to DMF solutions of TiCl4(THF)2 for 5 days at 85 °C in order to achieve PSE with Ti(IV).
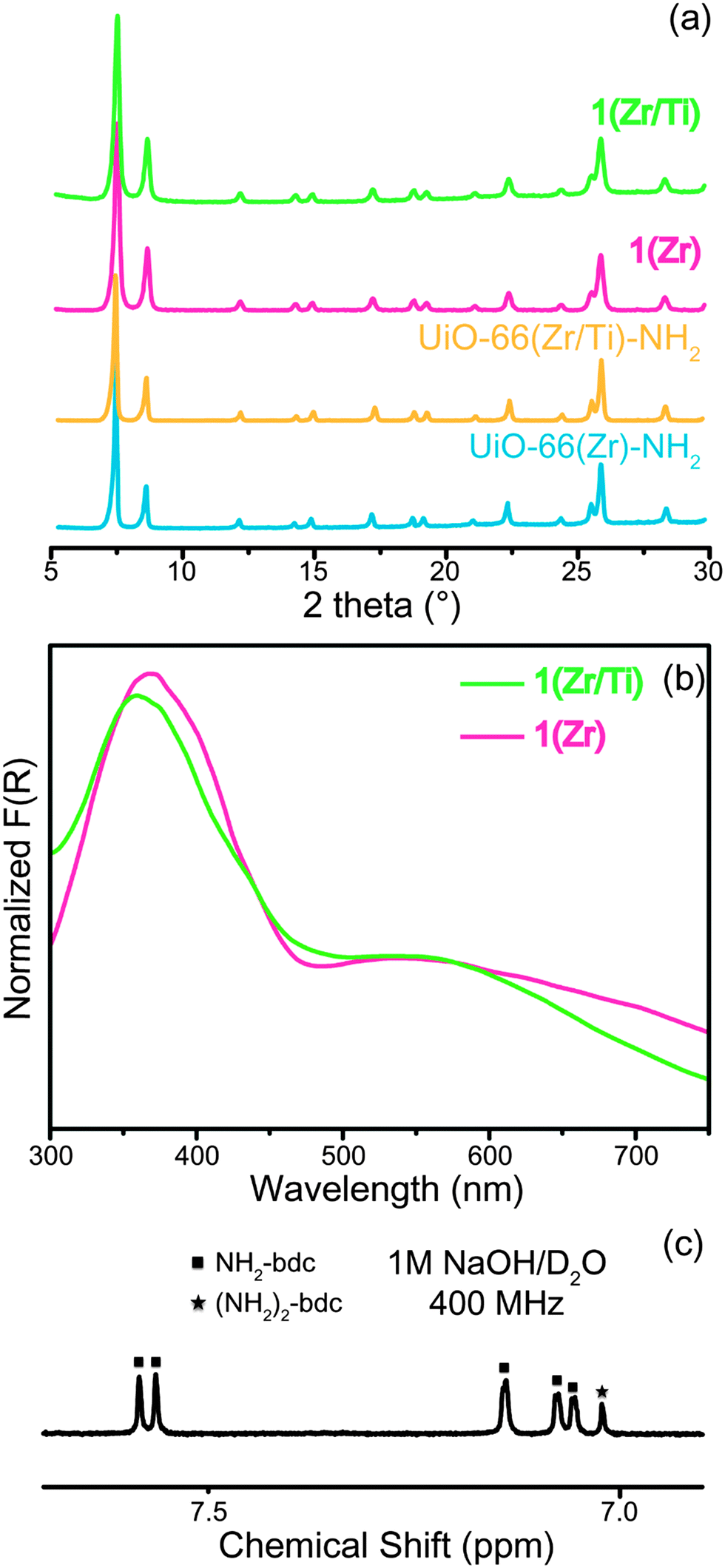 | ||
| Fig. 1 (a) PXRD patterns of UiO-66(Zr)-NH2, UiO-66(Zr/Ti)-NH2, 1(Zr), and 1(Zr/Ti). (b) Diffuse reflectance UV-visible spectra of 1(Zr) and 1(Zr/Ti) (calculated using the Kubelka–Munk function, F(R)). (c) 1H NMR of dissolved 1(Zr). | ||
After PSE, the crystallinity, morphology, and light absorption of the UiO-66 materials were maintained, indicating that PSE did not alter the gross physical properties of the MOFs. Introduction of Ti(IV) into the MOF was confirmed by both energy-dispersive X-ray spectroscopy (EDS) and inductively-coupled plasma mass spectrometry (ICP-MS) measurements. The ratios for Zr/Ti were 2.52 for 1(Zr/Ti) and 3.03 for UiO-66(Zr/Ti)-NH2 as determined by ICP-MS (Table S1, ESI†). The ICP-MS data support our argument that Ti(IV) was substituted for Zr(IV) in MOF SBUs. The weight percentage of both elements in 1(Zr/Ti) was determined to be 23.2 wt% for Zr and 4.8 wt% for Ti. If Ti was simply loaded into 1(Zr) with no change in the SBUs, then these values should be 29.2 wt% for Zr and 6.1 wt% for Ti (Table S2, ESI†). Furthermore, the surface area of 1(Zr) was essentially unchanged after PSE (from 937 ± 7 m2 g−1 to 1004 ± 9 m2 g−1), indicating that the added Ti is not blocking the pores of the MOF. These suggest that, on average, the Zr6 SBUs were converted by PSE to ∼Zr4.3Ti1.7 for 1(Zr/Ti) and ∼Zr4.5Ti1.5 for UiO-66(Zr/Ti)-NH2.
Both MOFs were tested for photocatalytic CO2 reduction under visible light irradiation. The reaction was conducted in 5 mL of a mixed solution of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) acetonitrile (MeCN)–triethanolamine (TEOA, as a sacrificial base), and 0.1 M 1-benzyl-1,4-dihydronicotinamide (BNAH, as a sacrificial reductant).27–29 The suspension, which contained 5 mg of MOF, was purged with 1 bar of CO2 gas for 30 min followed by light irradiation by a 300 W Xe lamp. The initial pH value of the 4:1 MeCN–TEOA solution was ∼11, but dropped to ∼9.5 after purging with CO2 gas. Because of the high pH, the photocatalysis products can be deprotonated; therefore, the reaction mixtures were extracted with ethyl acetate and washed with H2SO4 (to remove TEOA and protonated products).30 The final ethyl acetate solution (1 μL) was analyzed by gas chromatography-mass spectrometry (GC-MS) to identify the products.
:1 (v/v) acetonitrile (MeCN)–triethanolamine (TEOA, as a sacrificial base), and 0.1 M 1-benzyl-1,4-dihydronicotinamide (BNAH, as a sacrificial reductant).27–29 The suspension, which contained 5 mg of MOF, was purged with 1 bar of CO2 gas for 30 min followed by light irradiation by a 300 W Xe lamp. The initial pH value of the 4:1 MeCN–TEOA solution was ∼11, but dropped to ∼9.5 after purging with CO2 gas. Because of the high pH, the photocatalysis products can be deprotonated; therefore, the reaction mixtures were extracted with ethyl acetate and washed with H2SO4 (to remove TEOA and protonated products).30 The final ethyl acetate solution (1 μL) was analyzed by gas chromatography-mass spectrometry (GC-MS) to identify the products.
The photocatalysis products were analyzed via GC-MS, the results of which are shown in Fig. 2. 1(Zr/Ti) showed similar turnover number values over three photocatalytic cycles (6 hours each), indicating that the catalytic ability of the MOF was not degraded during photocatalysis. Turnover numbers were calculated based on the Ti content determined from ICP-MS results. The average turnover number of 6.27 ± 0.23 (31.57 ± 1.64 μmol of HCOOH, from 3 independent samples) indicates that each Ti site transferred about 13 electrons to CO2 over the course of each catalytic run (Fig. S3, ESI†). UiO-66(Zr/Ti)-NH2 gave a lower turnover number (4.66 ± 0.17) when compared to 1(Zr/Ti) (Fig. 2 and Fig. S4, ESI†). No HCOOH was detected when using the parent 1(Zr) and UiO-66(Zr)-NH2 materials, indicating that Ti was essential for photocatalysis.
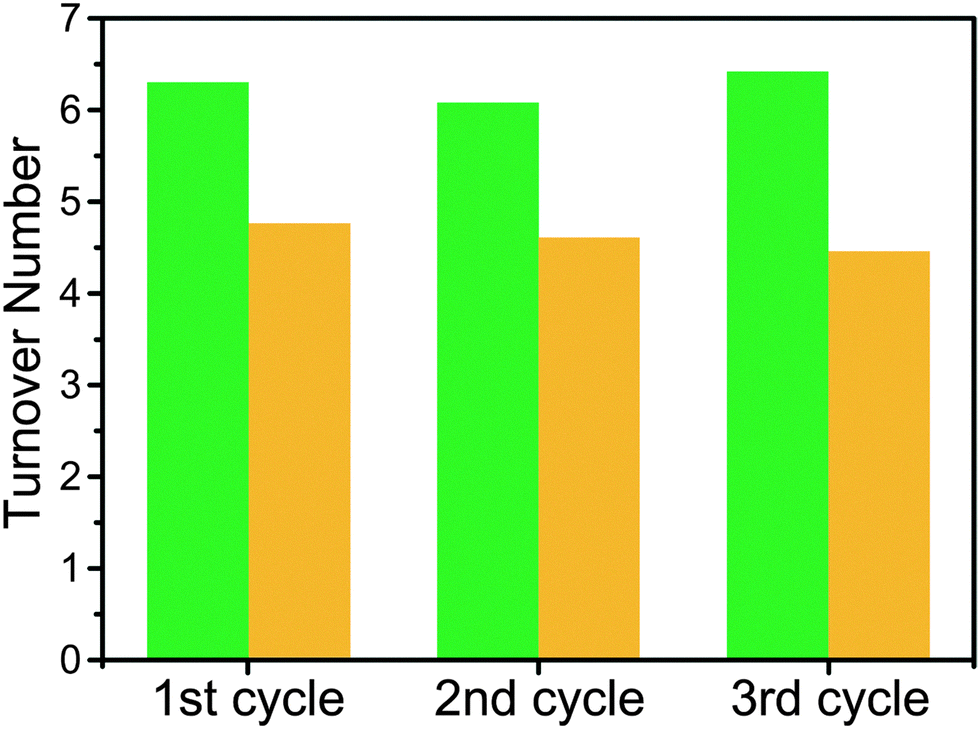 | ||
| Fig. 2 Turnover numbers of 1(Zr/Ti) (green) and UiO-66(Zr/Ti)-NH2 (gold) for photocatalysis of CO2 to HCOOH over three cycles. Samples were recovered after each cycle and reused under identical reaction conditions. | ||
The MOFs were studied by photoluminescence (PL) spectroscopy to provide evidence for charge transfer in 1(Zr/Ti). As shown in Fig. 3a, the emission intensity of 1(Zr) was remarkably reduced after PSE. This indicates that the recombination rate of photogenerated electron–hole pairs in the organic linkers was decreased suggesting that charges were transferred to the inorganic SBUs. In addition, 1(Zr/Ti) achieved better charge separation than UiO-66(Zr/Ti)-NH2 because 1(Zr/Ti) quenched a greater portion of the photogenerated charges than UiO-66(Zr/Ti)-NH2 (Fig. S5, ESI†). This indicates that 1(Zr/Ti) accepts more electrons from the organic linkers to catalyze CO2 than UiO-66(Zr/Ti)-NH2, which is supported by the GC-MS results.
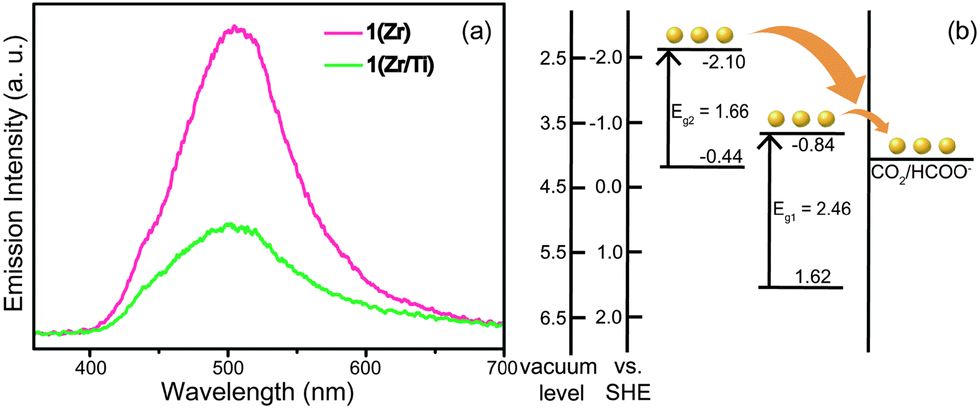 | ||
| Fig. 3 (a) Photoluminescence spectra of both 1(Zr) and 1(Zr/Ti). (b) Energy band structure of 1(Zr/Ti) derived from UPS and F(R) results. Heterogeneous ligands formed two energy levels in MOF, which potentially catalyze CO2. | ||
The energy band structures of both 1(Zr/Ti) and UiO-66(Zr/Ti)-NH2 were investigated using UV light photo-electron spectroscopy (UPS, Fig. S6, ESI†). The UPS results indicate that 1(Zr/Ti) has two different valence bands, in contrast to UiO-66(Zr/Ti)-NH2 that has a single valence band. This is consistent with the UV-visible spectra in Fig. 1(b) where 1(Zr/Ti) showed two absorption bands. The maxima of the two valence bands of 1(Zr/Ti) were calculated to be 1.62 eV and −0.44 eV (vs. SHE) from the UPS spectrum of 1(Zr/Ti) and bandgap energies for those two levels were determined from UV-vis spectra to be 2.46 eV and 1.66 eV, respectively. These data can be used to produce an energy band diagram of 1(Zr/Ti) as shown in Fig. 3(b). Two conduction band minima values of −0.84 eV and −2.10 eV (vs. SHE) are suitable for electron transfer to CO2.31 UiO-66(Zr/Ti)-NH2 has a single valence band maximum of 1.88 eV and its conduction band minimum was calculated to be −0.79 eV based on the bandgap energy for UiO-66(Zr/Ti)-NH2 of 2.67 eV (calculated from the UV-vis spectrum, Fig. S7, ESI†). Consequently, 1(Zr/Ti) is expected to show better photocatalytic efficiency than UiO-66(Zr/Ti)-NH2 because 1(Zr/Ti) has two light absorption routes (both suitable for CO2 reduction) originating from the (NH2)2-bdc ligand.
In order to prove that HCOOH is produced from photocatalytic reduction of CO2, the reaction was performed using 13CO2 and the products analyzed by 13C NMR. A solution containing CD3CN, TEOA, BNAH, and 13CO2 was subjected to identical photocatalysis conditions (see ESI†). Calibration of the expected product peaks was performed because the alkaline reaction conditions can perturb the chemical shifts of the products (Fig. S8, ESI†).30 H13COOH was found at 162.73 ppm in a CD3CN solution and this value was increased to 169.86 ppm due to deprotonation of H13COOH to H13COO− with added TEOA.
Several products, including H13COO−, 13CO2, H13CO32−, 13CO32−, residual solvent, and other small peaks were found in the 13C NMR spectrum after photocatalysis by both 1(Zr/Ti) after 13 hours of light irradiation (Fig. 4). 13CO32− and H13CO32− were produced by oxidation of 13CO2 under these alkaline conditions.31,32 Other small peaks come from isotopes of BNAH as these shifts were found in a reference solution of CD3CN–TEOA–BNAH (Fig. S8, ESI†). These results indicate that the carbon source of photocatalytically produced HCOOH is CO2 gas, and not other sources (such as MOF ligand decomposition). The 13C NMR spectrum of the product solution from UiO-66(Zr/Ti)-NH2 showed additional peaks in addition to BNAH and these peaks might represent organic linkers dissociated from the MOF during photocatalysis (Fig. S9, ESI†). No H13COO− was detected in NMR spectra when using 1(Zr) and UiO-66(Zr)-NH2, consistent with the GC-MS results.
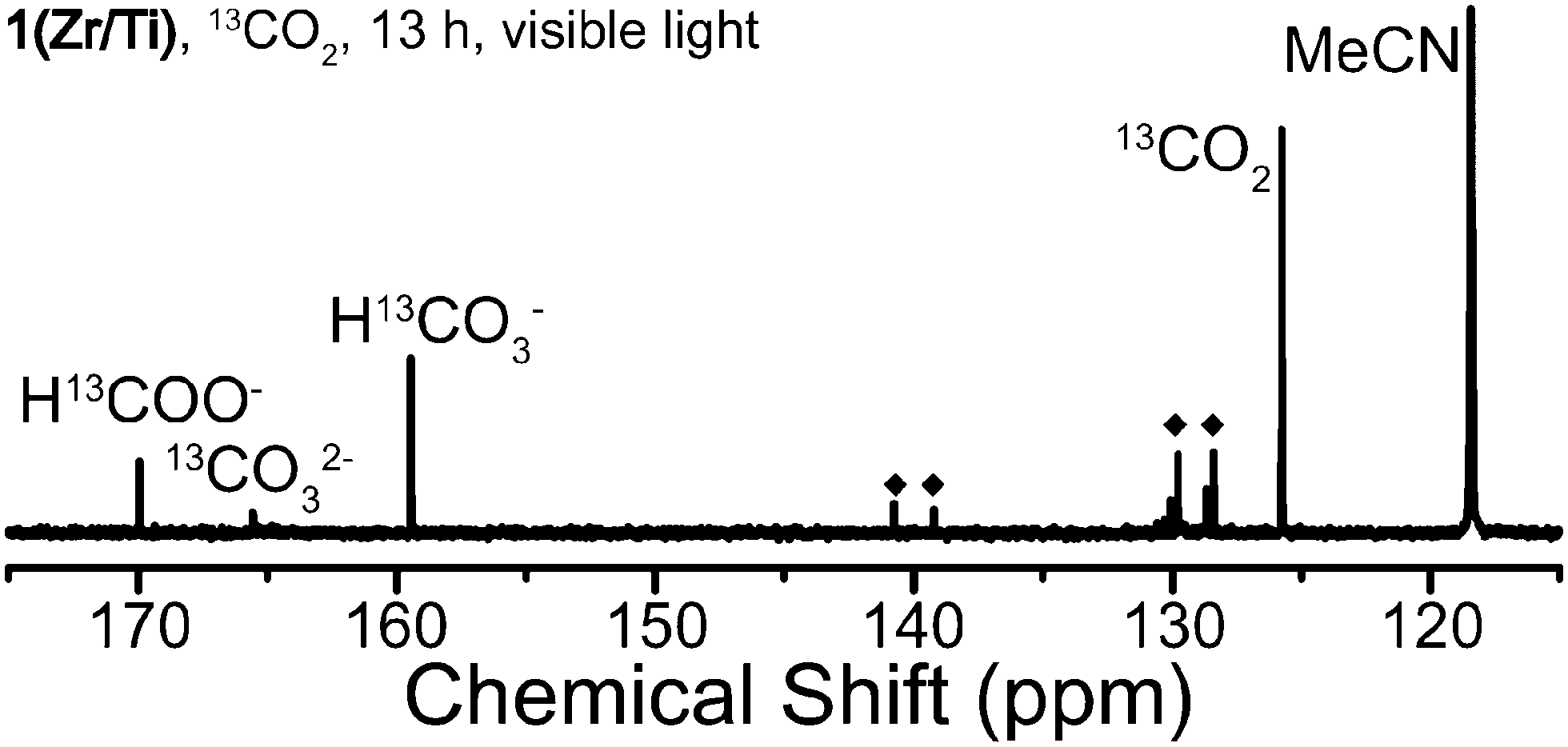 | ||
| Fig. 4 13C NMR spectrum of product solution from photocatalysis of 13CO2 by 1(Zr/Ti) for 13 hours under visible light irradiation. ◆ = BNAH. | ||
Durability is another important issue when considering long-term photocatalysis. The crystallinity, Zr/Ti ratio, and morphology of the UiO-66 materials were examined after three cycles of photocatalysis. Both 1(Zr/Ti) and UiO-66(Zr/Ti)-NH2 maintained the crystallinity and morphology after three cycles of photocatalysis (Fig. S10 and S11, ESI†). However, UiO-66(Zr/Ti)-NH2 showed broadened PXRD reflections, a roughened surface in SEM image, and greater leaching of Ti (as measured by ICP-MS, Table S1, ESI†), compared to 1(Zr/Ti). Overall, 1(Zr/Ti) was more stable than UiO-66(Zr/Ti)-NH2 during photocatalysis.
To evaluate our mixed-metal approach, the photocatalytic activity of NH2-MIL-125(Ti) was also investigated. NH2-MIL-125(Ti) was synthesized following a reported procedure and its structure confirmed by PXRD (Fig. S12, ESI†).13 Photocatalysis with NH2-MIL-125(Ti) under the same reaction conditions described above gave a turnover number of 1.52 after 6 hours. This value is much lower when compared to the MOFs described here. Therefore, it is inferred that the mixed metal SBUs described here are more efficient photocatalysts. This might be, in part, because the Ti8 SBUs have a redox potential that is too low to provide a sufficient driving force to catalyze CO2 reduction when compared to the mixed Zr/Ti SBUs described here.
The photocatalytic ability of 1(Zr/Ti) was also compared to heterogeneous photocatalytic systems based on semiconductors or MOFs in units of turnover frequency (h−1) (Tables S3 and S4, ESI†).33–381(Zr/Ti) prepared in this work showed a much higher photocatalytic ability, and also improved visible light sensitivity than non-MOF heterogeneous photocatalysts. In the comparison to MOF-based photocatalysts, only one MOF system we are aware of shows better catalytic ability than 1(Zr/Ti), but this system requires Ru for both the catalytic site and an exogenous light sensitizer.38 In the latter example, the reaction conditions are not identical to those reported here, which makes an absolute comparison of photoreactivity difficult.
In summary, highly efficient and robust MOF photocatalysts for CO2 reduction to HCOOH under visible light irradiation, without the need for an exogenous light sensitizer, were developed via PSE of Ti(IV) into a series of UiO-66 MOFs. UiO-66 materials were tuned to have catalytic activity by lowering the electron accepting levels of the Zr6−xTix (Zr6−xTixO4(OH)4) SBUs. Introduction of diamine-substituted ligands greatly enhanced the photocatalytic ability by introducing new energy levels for additional light absorption and charge transfer. This study suggests a new approach to develop MOF photocatalysts using a mixed-metal and mixed-linker approach.
The majority of the experiments described, including all of the synthetic work and MOF characterization, was supported by a grant from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under Award No. DE-FG02-08ER46519 (Y.L., S.M.C.). Additional support for S.K. and J.K.K. was provided by the Korea Center for Artificial Photosynthesis (KCAP) funded by the National Research Foundation of Korea (2009-0093881).
Notes and references
- H. D. Matthews, N. P. Gillett, P. A. Stott and K. Zickfeld, Nature, 2009, 459, 829–832 CrossRef CAS PubMed.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- K. Sumida, et al. , Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- Y. Fu, et al. , Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- Y. Horiuchi, et al. , J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- L. Valenzano, et al. , Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- Q. Yang, et al. , Chem. Commun., 2011, 47, 9603–9605 RSC.
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4106 RSC.
- M. Servalli, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 1904–1906 RSC.
- F. Vermoortele, et al. , J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- W. Liang, R. Babarao and D. M. D'Alessandro, Inorg. Chem., 2013, 52, 12878–12880 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- O. Karagiaridi, et al. , Chem. Sci., 2012, 3, 3256–3260 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- S. Takaishi, E. J. DeMarco, M. J. Pellin, O. K. Farha and J. T. Hupp, Chem. Sci., 2013, 4, 1509–1513 RSC.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- H. Fei, et al. , J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- C. Pac, M. Ihama, M. Yasuda, Y. Miyauchi and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 6495–6497 CrossRef CAS.
- X. Q. Zhu, et al. , Chem. – Eur. J., 2003, 9, 3937–3945 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- S. Moret, P. J. Dyson and G. Laurenczy, Dalton Trans., 2013, 42, 4353–4356 RSC.
- X.-H. Xia, et al. , Carbon, 2007, 45, 717–721 CrossRef CAS PubMed.
- T. Baran, S. Wojtyła, A. Dibenedetto, M. Aresta and W. Macyk, Appl. Catal., B DOI:10.1016/j.apcatb.2014.09.052.
- Q. Zhang, C.-F. Lin, Y. H. Jing and C.-T. Chang, J. Air Waste Manage. Assoc., 2014, 64, 578–585 CAS.
- G. Mele, et al. , Molecules, 2015, 20, 396–415 CrossRef CAS PubMed.
- L. Li, et al. , Chem. Sci., 2014, 5, 3808–3813 RSC.
- D. Sun, et al. , Chem. Commun., 2015, 51, 2645–2648 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, details of results. See DOI: 10.1039/c5cc00686d |
| This journal is © The Royal Society of Chemistry 2015 |