
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient direct 2,2,2-trifluoroethylation of indoles via C–H functionalization†‡
Gergely L.
Tolnai§
a,
Anna
Székely§
a,
Zita
Makó
a,
Tamás
Gáti
b,
János
Daru
c,
Tamás
Bihari
c,
András
Stirling
*c and
Zoltán
Novák
*a
aMTA-ELTE “Lendület” Catalysis and Organic Synthesis Research Group, Institute of Chemistry, Eötvös University, Faculty of Science, Pázmány Péter stny. 1/A, 1117, Budapest, Hungary. E-mail: novakz@elte.hu; Web: http://zng.elte.hu
bServier Research Institute of Medicinal Chemistry, Záhony utca 7, H-1031 Budapest, Hungary
cResearch Centre for Natural Sciences of the Hungarian Academy of Sciences, Department of Theoretical Chemistry, Magyar Tudósok körútja 2, 1117 Budapest, Hungary. E-mail: stirling.andras@ttk.mta.hu
First published on 6th February 2015
Abstract
A novel highly C3 selective metal free trifluoroethylation of indoles using 2,2,2-trifuoroethyl(mesityl)-iodonium triflate was developed. The methodology enables the introduction of a trifluoroethyl group in a fast and efficient reaction under mild conditions with high functional group tolerance. Beyond the synthetic developments, quantum chemical calculations provide a deeper understanding of the transformation.
The presence of fluorine in organic molecules often provides advantageous properties to the molecules, as fluorinated functional groups can beneficially modify the electronic properties of the compounds, improve their metabolic stability, and enhance their lypophilicity. Therefore the development of new synthetic methods for the installation of fluorine and fluorous functional groups is an emerging field in synthetic organic chemistry.1
Modification of lead compounds with fluorous functional groups to fine-tune their biological activity is one of the most frequently used strategies in medicinal chemistry. Aside from the simple fluorine atom and CF3 group, the presence of other related small fluorous functional groups such as trifluoromethoxy,2 trifluoromethylthio3 or structurally similar trifluoroethyl4 groups on an aromatic core structure has also gained exceptional attention from the fields of synthetic organic and medicinal chemistry.
Due to the importance of the indole framework in the structure of drug candidates much attention has been focused on the synthesis of fluoroalkylated indole derivatives. In general, trifluoroethylated indoles are accessible through indole core functionalization via multistep syntheses (3–6 synthetic steps)5 or ring constructions such as a novel radical Fisher indole type synthesis developed by Antonchick.6 Compared to trifluoromethylation, the synthetic transformations to access trifluoroethylated compounds via C–C bond formation are underdeveloped. The main, but still uncommon methods for the construction of trifluoroethyl groups involve the trifluoromethylation of a benzylic position,7 the addition of a trifluoromethyl group to alkenes,8 and the utilization of gaseous trifluoromethyl diazomethane.9 Although the unique electronic properties of the CF3CH2X (X = Br, I, OTf) compounds limit their use as simple electrophiles,10 recently several cross-coupling approaches were developed for the introduction of trifluoroethyl groups into aromatic systems.11 The direct C–H functionalization with a trifluoroethyl group is seldom explored. Very recently Ackermann described the first nickel catalyzed trifluoroethylation process utilizing an 8-aminoquinoline directing group and trifluoroethyliodide as alkyl sources.12 A Catellani type palladium catalyzed cascade trifluoroethylation was developed by Liu for the synthesis of olefinated trifluoroethyl arenes.13 A unique example for transition metal catalyst free direct trifluoroethylation of heterocycles is Baran’s radical alkylation strategy using zinc sulfinate salts.14
In our work, we aimed to develop a new alkylation methodology for the direct trifluoroethylation of indoles with the aid of an electrophilic trifluoroethyl synthon originated from hypervalent iodonium salts. Introduction of hypervalent iodonium salts as useful reagents in organic chemistry15 opens new possibilities for the formation of carbon–heteroatom and carbon–carbon bonds via C–H activation.16 Besides numerous synthetic applications,15a,17 the use of λ3 iodo compounds provides an emerging transition metal catalyzed strategy for the direct functionalization of indoles.18 The pioneering work of Umemoto in the field of fluoroalkyliodonium salts19 inspired us to use hypervalent iodine reagents for the trifluoroethylation of indoles. These reagents were mostly used for the fluoroalkylation of heteroatom20 and soft carbon21 nucleophiles to build C–N, C–O, C–S and C(sp3)–C(sp3) bonds.
For the realization of the desired transformation we synthesized the 2,2,2-trifuoroethyl(mesityl)-iodonium triflate salt (1a) as a new reagent from bis(trifluoroacetatoxy)(2,2,2-trifluoroethyl)iodide and mesitylene in 94% isolated yield on a multigram scale. We strategically chose the mesityl group (Mes) as a frequently used aromatic system for the synthesis of aryliodonium salts with high stability and activity. However, two additional trifluoroethylaryliodonium salts were prepared, but the phenyl (1b) and 4-tolyl (1c) derivatives were obtained with lower efficiency (68% and 83% respectively).
After having the designed reagents in hand, we turned our attention to the possible C–H functionalization of unprotected indoles to develop a powerful synthetic methodology to enable C–C bond formation without the necessity for protecting groups. First, we studied the trifluoroethylation reaction of indole (2a) with 1a in dichloromethane (CH2Cl2) at 25 °C with or without transition metal catalysts (Cu(II), Pd(II), and Ag(I) salts). To our delight the reaction of unsubstituted indole provided the desired C3 trifluoroethylated product 3a (Scheme 1). In contrast to the known iodonium salt based transition metal catalyzed indole functionalization methodologies, we found that the presence of transition metal catalysts had deleterious effects on the trifluoroethylation reaction.22
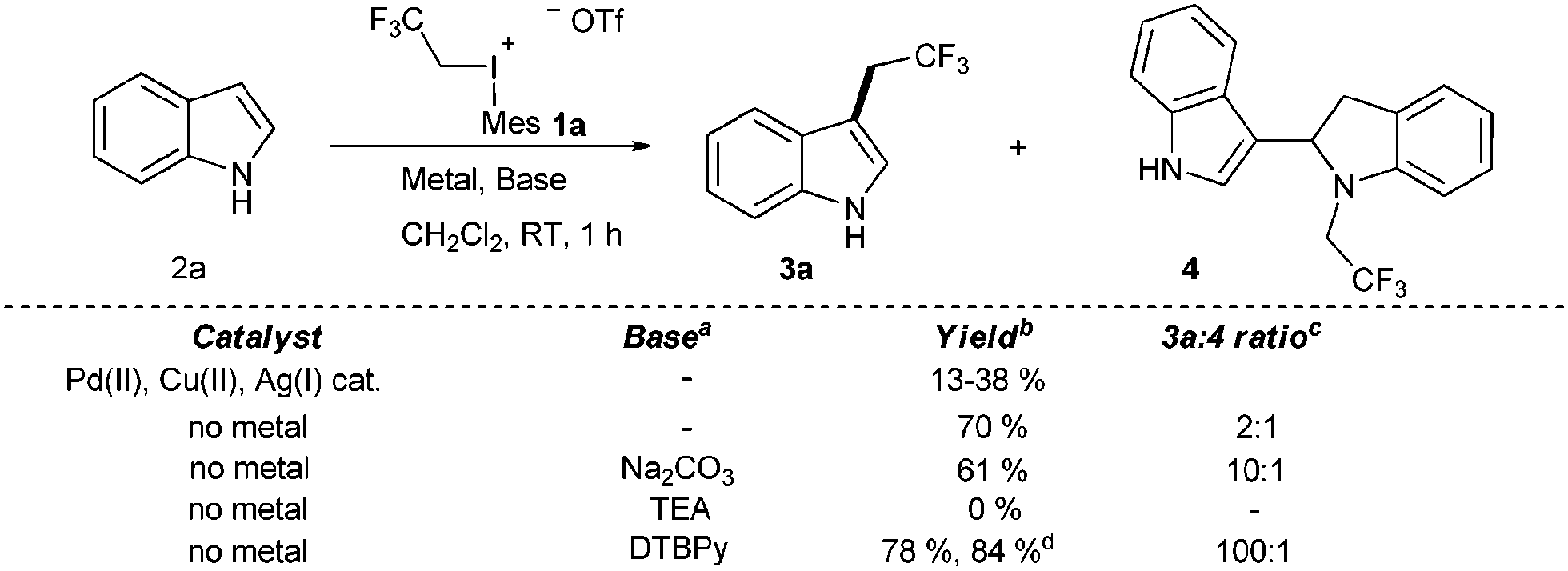 | ||
| Scheme 1 Adjustment of reactivity by different bases. a 2 equivalents of the base, 1 equivalent of the 1a reagent. b Isolated yield of the mixture. c Determined by 19F NMR measurements. d 1.3 equivalent of the iodonium salt (1a) was used. | ||
Despite the high reactivity of the polyfluoroalkyl λ3 iodonium compounds toward heteroatom nucleophiles20a,b trifluoroethylation did not occur on the indole nitrogen due to its lower nucleophilicity. Besides these beneficial features, we observed the formation of a significant amount of N-trifluoroethylated indolino-indole derivative (4). The presence of trifluoromethanesulfonic acid, generated in the reaction, accelerates the dimerization of indole, and the formed indoline moiety undergoes straightforward N-trifluoroethylation.22 It was anticipated that the amount of this side product could be decreased by adding an appropriate base such as 2,6-di-tert-butylpyridine (DTBPy) and target compound 3a was isolated in 84% yield.22 Next, we explored the synthetic utility of this reaction by examining the substrate scope in CH2Cl2 at 25 °C.22 Indole and alkyl indoles reacted smoothly with 1a under our conditions to provide trifluoroethyl indoles 3a–3e in 10 minute reaction time (Scheme 2). In the case of 3-methylindole, formation of any trifluoroethylated product were not observed (not shown). More electron rich alkoxyindoles were also successfully functionalized in a rapid reaction (10–60 minutes) and the desired compounds (3f–j) could also be isolated in good to excellent yields (46–96%). Although, the indole nitrogen does not require protection, the tested N-methyl and N-TIPS derivatives were also successfully trifluoroethylated (3k–m). Nucleophilic functional groups connected to the indole frame such as free hydroxyl, amino and carboxyl functions could be masked with different protecting groups for the efficient trifluoroethylation (3n–p) of indole at position 3 to obtain valuable heterocyclic building blocks. Indole containing TMS protected alkyne was also converted to its corresponding trifluoroethylated compound (3q) in 84% yield, opening the way for further synthetic transformations through the acetylene part. To further explore the real synthetic power of the method, we examined the scope with electron withdrawing groups on the indole skeleton. To our delight the presence of fluorine, nitro, cyano and trifluoromethyl groups on the indole core was well tolerated, and the desired products (3r–v) were isolated in 31–94% yield after 1.5–4 hours of reaction time. Demonstrating the power of the methodology we examined the trifluoroethylation of indoles bearing functional groups suitable for cross coupling chemistry (Cl, Br, I, and boronic ester; Scheme 3). Each halogen substituted indole gave the desired products (3w–3ee) in good to excellent yields (61–90%), as well as the boronic ester derivative (3ff, 83%).
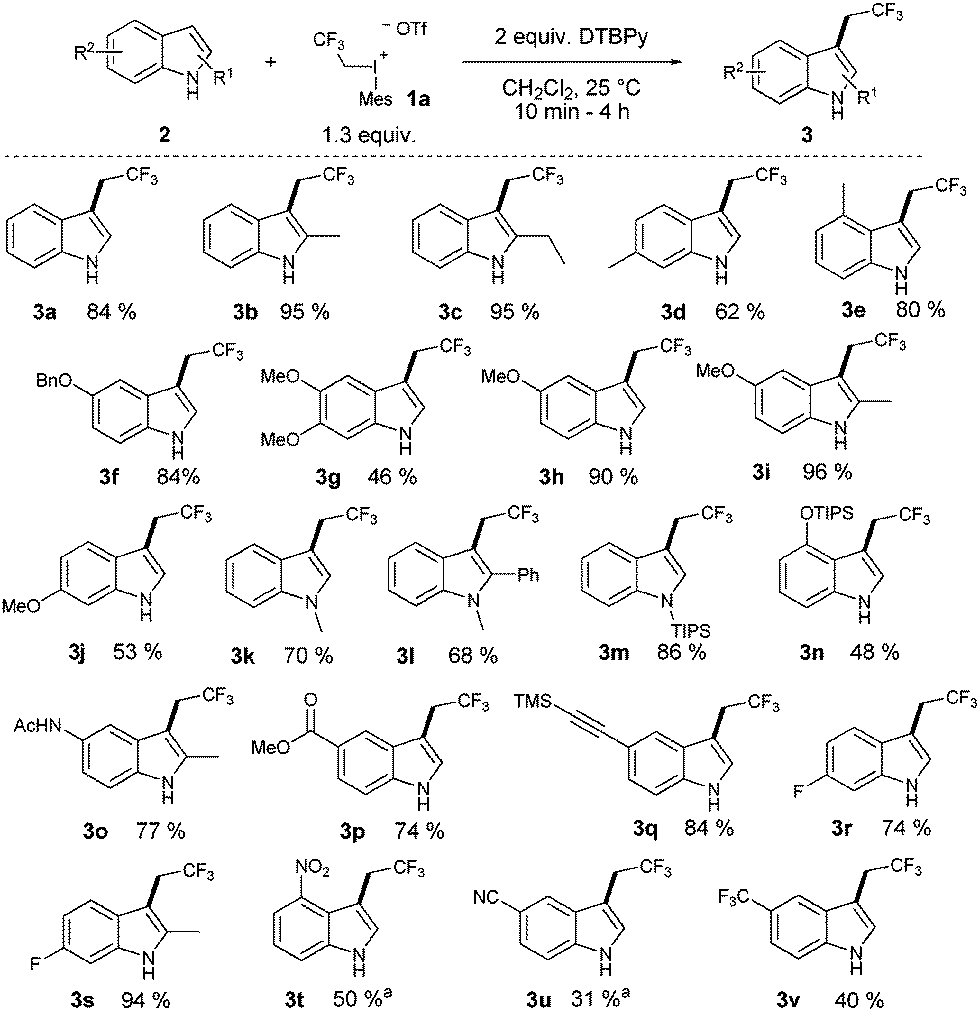 | ||
| Scheme 2 Substrate scope of the transformation: a without base, 2 equiv. of the iodonium salt. | ||
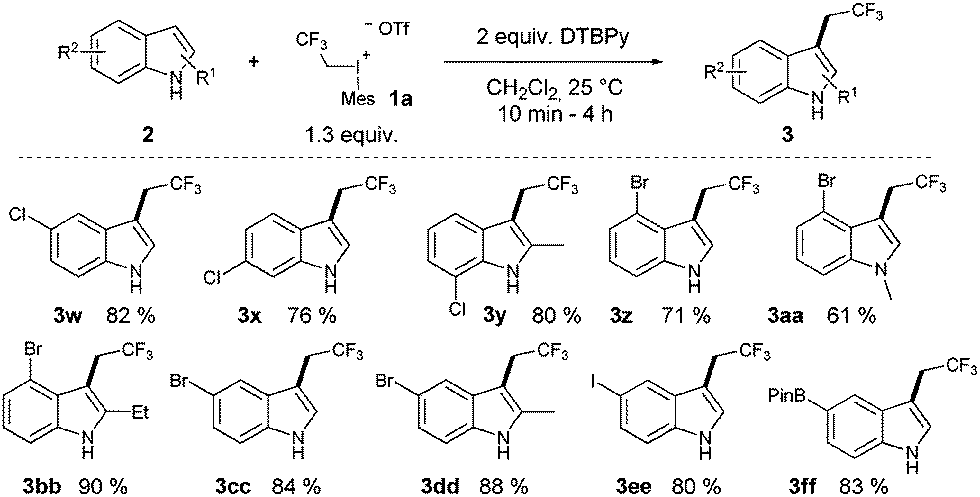 | ||
| Scheme 3 Trifluorethylated indoles for cross-coupling chemistry. | ||
We employed DFT calculations to understand the mechanism and the selectivity of the present reaction. First we considered the reaction between indole (2a) and 2,2,2-trifluoroethyl(mesityl)-iodonium triflate (1a) (Scheme 4). The dissociation of the triflate anion proved to be slightly exergonic (−0.9 kcal mol−1) suggesting a dissociated resting state. The rate-determining step is the trifluoroethylation of the indole-ring. The next step is the deprotonation of the σ-complex by the basis with a 18.3 kcal mol−1 barrier. Both steps are highly exergonic indicating irreversible transformations. Formation of 1- and 2-substituted indoles is much less favourable, which explains that they could not be observed in experiment.22 We have also calculated the free energy barrier heights for the all the substrates indicated in Schemes 2 and 3.22 Within the expected accuracy the calculations nicely correlate with experiment providing confidence in the conclusion. The formation of 3-indolyl-trifluorethyl-mesityl-iodonium salt from 1a and 2a as another possible intermediate of the reaction and subsequent product formation through reductive elimination have also been taken into consideration. This reaction pathway has been safely excluded on the basis of the prohibitively high barrier (52 kcal mol−1) for the formation of the key intermediate from 1a and 2.
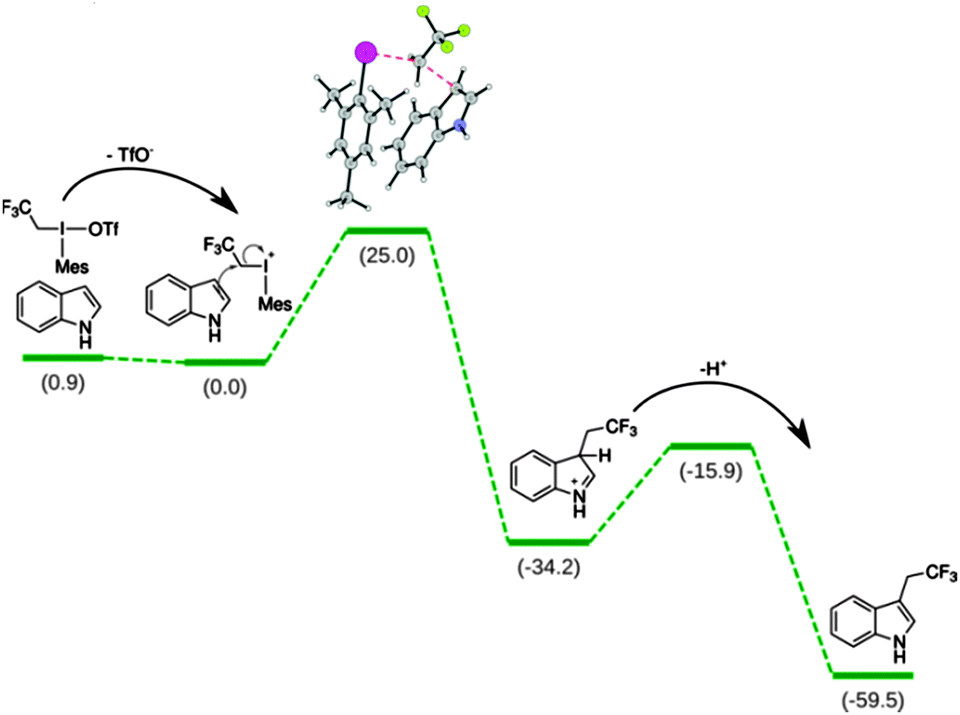 | ||
| Scheme 4 Free energy profile in kcal mol−1 of the transformation based on DFT calculations. | ||
To explain the special efficiency of DTBPy as a base in the reaction we have also calculated the activation barriers of the alkylations of different amine bases such as TEA, DIPEA, 2,6-lutidine and DTBPy. It was found that the activation energies of the N-trifluoroethylation of these bases with 1a were 18.4 kcal mol−1, 22.4 kcal mol−1 and 23.6 kcal mol−1, respectively, which are in the same range as the barriers of the C alkylations of the studied indoles. However, computation results showed that the N-trifluoroethylation of DTBPy required an extremely high activation energy (36.2 kcal mol−1) due to the significant steric repulsion.22 These results indicate that variations in the barrier heights of trifluoroethylations can strongly influence the outcome of the reaction, which is important from a synthetic standpoint. Additional experiments have convincingly confirmed this prediction as shown in Scheme 5. In this scheme we collected the conversions of four indole derivatives in the presence of different bases. From left to right the reactivity (TS barriers of trifluoroethylation) of the indole derivatives are tuned by varying their substituents. Going down in the columns, the barriers of N-alkylation increase. The trends obtained for the all possible combinations of indole–base pairs demonstrate that the efficiency of a substrate–base combination is determined by the activation barrier differences of the competing N- and C-alkylations. Clearly, alkylations of the bases and substrates are competitive reactions, and the yields support the postulated kinetic control.
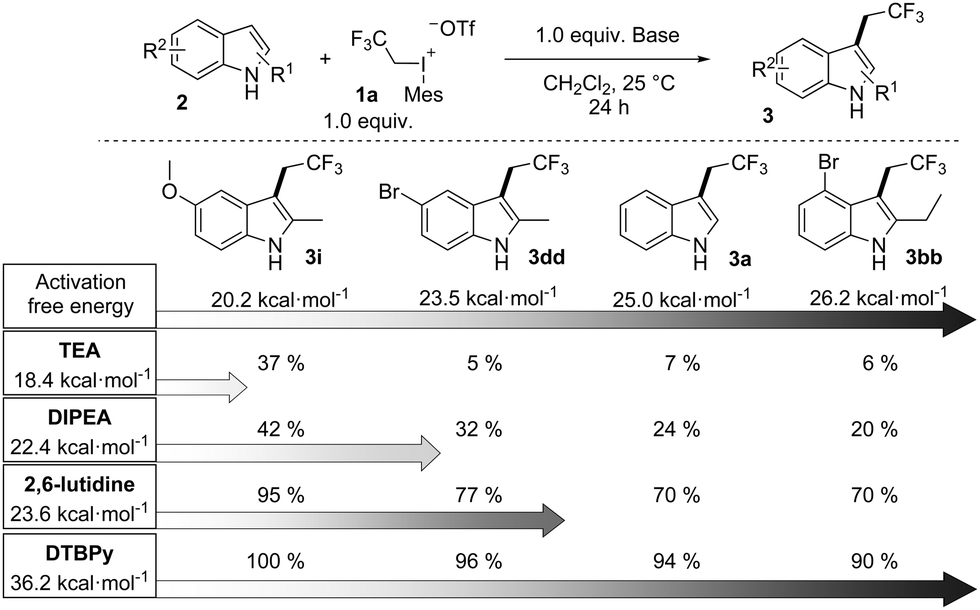 | ||
| Scheme 5 GC Conversion of trifluoroethyl indole derivatives obtained from reactions carried out in the presence of different amines. Activation energies of trifluoroethylation of indole derivatives and bases are indicated. | ||
In conclusion, we have developed a transition metal free direct C–H trifluoroethylation reaction, which enables the selective trifluoroethylation of a heterocyclic system for the first time resulting in C(sp2)–C(sp3) bond formation.
The readily accessible new 2,2,2-trifluoroethyl(mesityl)-iodonium triflate reagent ensures the straightforward trifluoroethylation of indoles at position 3 under very mild conditions in a rapid reaction (10–240 minutes of reaction time). The excellent functional group tolerance of the developed transformation enables the access of chemically diverse compound classes with potential medicinal interest. Additionally, with the aid of DFT studies we revealed the mechanistic steps of the reaction, and explained the important role of basic additives in the transformation process. Beyond these mechanistic studies and the synthesis of 3-trifluoroethyl indoles with high versatility the developed methodology opens new doors to the synthesis of other trifluoroethylated aromatic and heterocyclic compounds via C–H functionalization or transition metal catalyzed C–H activation. The study of these synthetic possibilities is currently undergoing in our laboratory.
This project was supported by the “Lendület” Research Scholarship of the Hungarian Academy of Sciences (LP2012-48/2012), TÉT-10-1-2011-0245 and by OTKA K-101115. The authors thank Brian M. Stoltz at Caltech for proofreading this manuscript. Generous donation of indole derivatives by Servier Research Institute of Medicinal Chemistry is also acknowledged. Computational resources provided by the NIIF Supercomputer Center are acknowledged.
Notes and references
- (a) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201410288; (b) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432 CrossRef PubMed; (d) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (e) A. Harsanyi and G. Sandford, Green Chem., 2015 10.1039/C4GC02166E; (f) Z. Gonda, S. Kovács, C. Wéber, T. Gáti, A. Mészáros, A. Kotschy and Z. Novák, Org. Lett., 2014, 16, 4268 CrossRef CAS PubMed.
- (a) C. Huang, T. Liang, S. Harada, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 13308 CrossRef CAS PubMed; (b) O. Marrec, T. Billard, J.-P. Vors, S. Pazenok and B. R. Langlois, Adv. Synth. Catal., 2010, 352, 2831 CrossRef CAS; (c) F. Venturini, W. Navarrini, A. Famulari, M. Sansotera, P. Dardani and V. Tortelli, J. Fluorine Chem., 2012, 140, 43 CrossRef CAS PubMed.
- (a) R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 1650 CrossRef CAS PubMed; (b) A. Tlili and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 6818 CrossRef CAS PubMed; (c) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, J. Am. Chem. Soc., 2013, 135, 8782 CrossRef CAS PubMed; (d) E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125 CrossRef CAS PubMed; (e) G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Goossen, Chem. Sci., 2014, 5, 1312 RSC; (f) J. Xu, X. Mu, P. Chen, J. Ye and G. Liu, Org. Lett., 2014, 16, 3942 CrossRef CAS PubMed; (g) X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, Org. Lett., 2014, 16, 2192 CrossRef CAS PubMed; (h) A. Harsányi, É. Dorkó, Á. Csapó, T. Bakó, C. Peltz and J. Rábai, J. Fluorine Chem., 2010, 132, 1241 CrossRef PubMed; (i) S. Alazet, L. Zimmer and T. Billard, Chem. – Eur. J., 2014, 20, 8589 CrossRef CAS PubMed; (j) C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316 CrossRef CAS PubMed.
- (a) M. D. Krasowski, Neuropharmacology, 2000, 39, 1168 CrossRef CAS; (b) N. Kumar, L. A. Solt, J. J. Conkright, Y. Wang, M. A. Istrate, S. A. Busby, R. D. Garcia-Ordonez, T. P. Burris and P. R. Griffin, Mol. Pharmacol., 2010, 77, 228 CrossRef CAS PubMed; (c) T. W. Ho, L. K. Mannix, X. Fan, C. Assaid, C. Furtek, C. J. Jones, C. R. Lines and A. M. Rapoport, Neurology, 2008, 70, 1304 CrossRef CAS PubMed.
- (a) T. Konno, J. Chae, T. Ishihara and H. Yamanaka, J. Org. Chem., 2004, 69, 8258 CrossRef CAS PubMed; (b) P. Stjernlöf, T. Elebring, J. Nilsson, B. Andersson, S. Lagerkvist, K. Svensson, A. Ekman, A. Carlsson and H. Wikström, J. Med. Chem., 1994, 37, 3263 CrossRef; (c) Y. Qiao, T. Si, M. H. Yang and R. A. Altman, J. Org. Chem., 2014, 79, 7122 CrossRef CAS PubMed; (d) A. Hall, A. Billinton, S. H. Brown, A. Chowdhury, G. M. Giblin, P. Goldsmith, D. N. Hurst, A. Naylor, S. Patel, T. Scoccitti and P. J. Theobald, Bioorg. Med. Chem. Lett., 2008, 18, 2684 CrossRef CAS PubMed.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 11960–11964 CrossRef CAS PubMed.
- (a) H. Kawai, T. Furukawa, Y. Nomura, E. Tokunaga and N. Shibata, Org. Lett., 2011, 13, 3596 CrossRef CAS PubMed; (b) Y. Miyake, S. Ota, M. Shibata, K. Nakajima and Y. Nishibayashi, Org. Biomol. Chem., 2014, 12, 5594 RSC; (c) N. V. Kondratenko, E. P. Vechirko and L. M. Yagupolskii, Synthesis, 1980, 932 CrossRef CAS; (d) L. Zhu, S. Liu, J. T. Douglas and R. A. Altman, Chem. – Eur. J., 2013, 19, 12800–12805 CrossRef CAS PubMed; (e) G. K. S. Prakash, P. V. Jog, P. T. D. Batamack and G. A. Olah, Science, 2012, 338, 1324 CrossRef CAS PubMed; (f) G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600 CrossRef CAS PubMed.
- (a) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed; (b) J. Xu, Y. Fu, D.-F. Luo, Y.-Y. Jiang, B. Xiao, Z.-J. Liu, T.-J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed; (c) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 CrossRef CAS PubMed; (d) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef CAS PubMed.
- (a) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 9085 CrossRef CAS PubMed; (b) C.-B. Liu, W. Meng, F. Li, S. Wang, J. Nie and J.-A. Ma, Angew. Chem., Int. Ed., 2012, 51, 6227 CrossRef CAS PubMed.
- (a) N. Bodor, M.-J. Huang, C. Szántay Jr. and C. Szántay, Tetrahedron, 1992, 48, 5823 CrossRef CAS; (b) K. Tanaka, T. Nakai and N. Ishikawa, Chem. Lett., 1979, 175 CrossRef CAS.
- (a) Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 1033 CrossRef CAS PubMed; (b) A. Liang, X. Li, D. Liu, J. Li, D. Zou, Y. Wu and Y. Wu, Chem. Commun., 2012, 48, 8273 RSC; (c) F. Leng, Y. Wang, H. Li, J. Li, D. Zou, Y. Wu and Y. Wu, Chem. Commun., 2013, 49, 10697 RSC; (d) L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634 CrossRef CAS PubMed; (e) Y.-S. Feng, C.-Q. Xie, W.-L. Qiao and H.-J. Xu, Org. Lett., 2013, 15, 936 CrossRef CAS PubMed; (f) J. Hwang, K. Park, J. Choe, H. Min, K. H. Song and S. Lee, J. Org. Chem., 2014, 79, 3267 CrossRef CAS PubMed.
- W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477–2480 CrossRef CAS PubMed.
- H. Zhang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 10174–10178 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed.
- (a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (d) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (e) V. V. Zhdankin, Hypervalent Iodine Chemistry, Wiley, Chichester, 2014 Search PubMed.
- (a) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- (a) A. Székely, Á. Sinai, E. B. Tóth and Z. Novák, Synthesis, 2014, 1871 Search PubMed; (b) Á. Sinai, Á. Mészáros, T. Gáti, V. Kudar, A. Palló and Z. Novák, Org. Lett., 2013, 15, 5654 CrossRef PubMed; (c) F. Zhang, S. Das, A. J. Walkinshaw, A. Casitas, M. Taylor, M. G. Suero and M. J. Gaunt, J. Am. Chem. Soc., 2014, 136, 8851 CrossRef CAS PubMed; (d) B. S. L. Collins, M. G. Suero and M. J. Gaunt, Angew. Chem., Int. Ed., 2013, 52, 5799 CrossRef CAS PubMed; (e) Q. Y. Toh, A. McNally, S. Vera, N. Erdmann and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 3772 CrossRef CAS PubMed; (f) A. J. Walkinshaw, W. Xu, M. G. Suero and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 12532 CrossRef CAS PubMed.
- (a) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (b) J. P. Brand, C. Chevalley and J. Waser, Beilstein J. Org. Chem., 2011, 7, 565–569 CrossRef CAS PubMed; (c) G. L. Tolnai, S. Ganss, J. P. Brand and J. Waser, Org. Lett., 2013, 15, 112 CrossRef CAS PubMed; (d) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed; (e) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS PubMed.
- T. Umemoto and Y. Gotoh, J. Fluorine Chem., 1985, 28, 235 CrossRef CAS.
- (a) T. Umemoto and Y. Gotoh, Bull. Chem. Soc. Jpn., 1991, 64, 2008 CrossRef CAS; (b) V. Montanari and K. Kumar, J. Am. Chem. Soc., 2004, 126, 9528 CrossRef CAS PubMed; (c) V. Montanari and G. Resnati, Tetrahedron Lett., 1994, 35, 8015 CrossRef CAS.
- (a) T. Umemoto and Y. Gotoh, Bull. Chem. Soc. Jpn., 1987, 60, 3823 CrossRef CAS; (b) T. Umemoto and Y. Gotoh, J. Fluorine Chem., 1986, 31, 231 CrossRef CAS.
- For further data and detailed optimization results see ESI‡.
Footnotes |
| † This paper is dedicated to Prof. József Rábai on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization of products and the details of DFT calculations. See DOI: 10.1039/c5cc00519a |
| § Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |