
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of an open-cage fullerene encapsulating hydrogen fluoride†
Andrea
Krachmalnicoff
,
Richard
Bounds
,
Salvatore
Mamone
,
Malcolm H.
Levitt
,
Marina
Carravetta
and
Richard J.
Whitby
*
Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: rjw1@soton.ac.uk
First published on 18th February 2015
Abstract
The first encapsulation of hydrogen fluoride in an open-cage fullerene is reported. Solution and solid-state NMR spectra of the novel open-cage endofullerene are described.
Open-cage fullerenes provide the opportunity of trapping atoms and molecules inside an inert three-dimensional environment – a molecular-scale “nanolaboratory”.1–3 Their supramolecular structure makes the study of effectively isolated molecules practically feasible over a wide range of conditions.4 Physical phenomena such as spin-isomer conversion have been observed inside fullerene cages.5 The incorporation of the small molecules H2,6–10 H2O,10–15 N2,15,16 CO,16,17 NH3,18 and CH4,19 inside the cavity of open-cage fullerenes has been reported. Completion of “Molecular Surgery” to reform the pristine C60 cage has been achieved only for H2@C60,7,10 and H2O@C60.10,14 Hydrogen fluoride (HF) is one of the most studied molecules, both theoretically and experimentally.20 Empirical studies on isolated HF molecules are complicated by its high chemical reactivity and its marked tendency to form aggregates and strong hydrogen-bonds to Lewis bases. Many experiments show the constant presence of oligomers, even in the gas phase.21 Enclosure in a fullerene would provide a constrained non-coordinating environment for a single molecule of HF and potentially allow novel studies on its spectral properties, particularly at cryogenic temperatures. Although the fully enclosed and symmetric HF@C60 structure would be ideal, open-cage fullerene hosts could provide many of the same advantages. Important physical properties such as ferroelectricity have been anticipated for endofullerenes enclosing freely-rotating molecules with an electric dipole moment.22
Herein we describe the insertion of HF into the cavity of open-cage fullerene 1, and the solution and solid-state NMR study of endofullerene HF@1.
Density Functional Theory (DFT) calculations gave activation energies of 64.3, 52.2. and 29.8 kJ mol−1 respectively for the entry of H2, H2O and HF into 2.23 The lower barrier for the incorporation of H2O compared to H2, though surprising, agrees with the experimental evidence10 (temperatures of 100 and 120 °C are required respectively) and is presumably due to attractive dipolar interactions between the polar molecule and the open fullerene neck. The very low activation energy predicted for the entry of HF into 2 and the possibility of trapping the endohedral HF via hemiacetal formation made HF@1 a viable target. The calculated binding energy for HF inside 2 (25.9 kJ mol−1) and the barrier for the release of HF from HF@2 (55.7 kJ mol−1) suggested that the loss of HF from HF@2 would be slow at room temperature allowing HF@1 to form.
The above calculations prompted us to attempt the filling of 2 with HF. Gaseous hydrogen fluoride forms relatively stable polymeric adducts with several organic bases;24 therefore we selected such compounds as a convenient source of anhydrous hydrogen fluoride. When a solution of tetra-ketone 2 or of its hydrate 1 was treated with a large excess of 70% w/w hydrogen fluoride in pyridine (HF-Py), HF@1 was isolated after basic work-up and chromatography (Scheme 1).
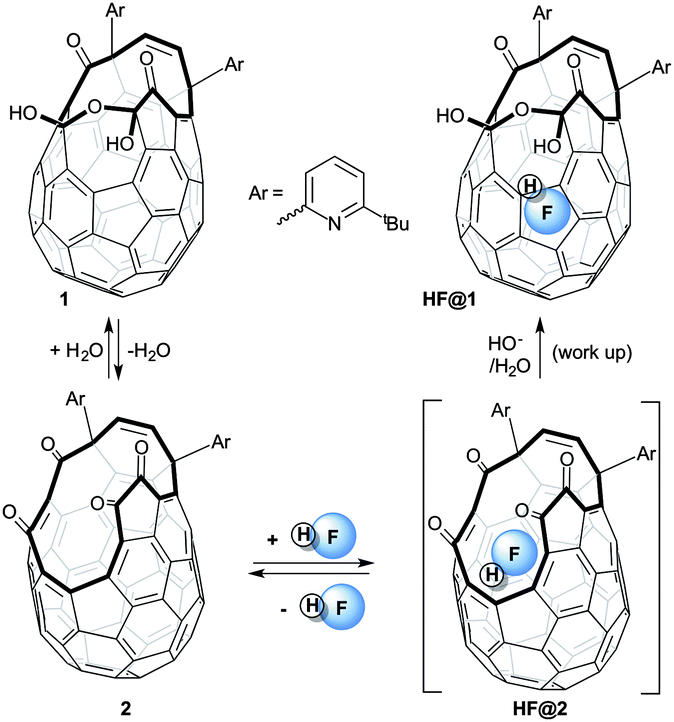 | ||
| Scheme 1 Generation of HF@1 from compound 1. | ||
The filling factor of HF@1 was established by 1H NMR spectroscopy, comparing the integral values of the endohedral HF proton with the protons on the exohedral groups. The highest filling factor (50%) was achieved by equilibrating a solution of 1 or 2 in dichloromethane with an excess (200 eq. HF) of HF-Py at room temperature. In these conditions the equilibrium was reached within 24 hours and prolonged reaction time did not afford a higher filling factor; 50% filled HF@1 was isolated after work up and chromatography in 89% yield. A larger excess (300 eq. HF) of HF-pyr did not increase the filling factor, but a lower excess (100 eq. HF) gave only 40% filled HF@1. Both compounds 1 and 2 can be used as substrate, as the acidic reaction medium is evidently able to afford the dehydration of 1 to form 2. The filling at 4 °C proceeded at a much slower rate (∼6 days) and did not improve the filling factor. At 80 °C a lower filling factor was obtained, as would be expected for an entropically disfavoured process, and decomposition of the substrate occurred giving 30% filled HF@1 in slightly lower isolated yield (80%). When the filling was carried out in non-chlorinated solvents such as benzene a biphasic system was formed and the substrate was quantitatively extracted into the HF-Py layer. After work-up and chromatography, 50% filled HF@1 was isolated in very good yield (88%). The ESI + MS spectrum of the isolated compounds displays signals at m/z 1121 and 1141 respectively for the molecular ions [1 + H]+ and [HF@1 + H]+.
Solution state NMR typically reveals unusual chemical shifts for the endohedral nuclei due to the strong magnetic shielding effect of the fullerene cage.4,25 Indeed the 1H signal from the HF molecule in HF@1 appears as a doublet centred at δ = −6.55 ppm with a JHF of 508 Hz (Fig. 1). This chemical shift is similar to those reported for the endohedral protons of H2@1 and H2O@1 which resonate at δ −7.17 and −9.84 ppm respectively.10,14 The large JHF value is comparable to that reported for HF in the gas phase.26
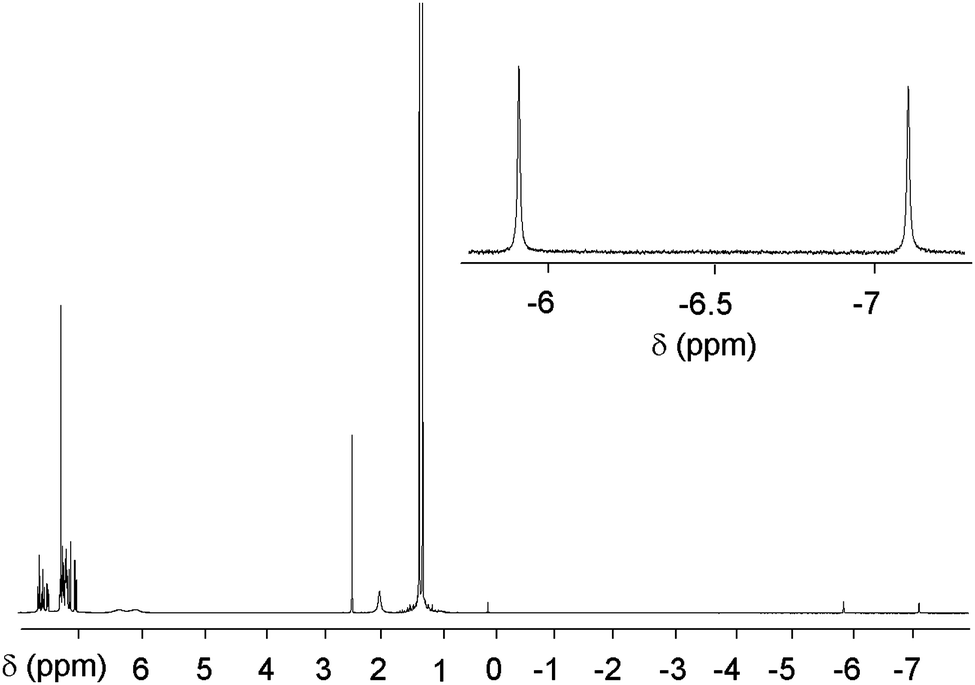 | ||
| Fig. 1 1H NMR spectrum (400 MHz, CDCl3) of HF@1. The δ scale is referenced to CHCl3. | ||
A doublet with a JHF of 508 Hz is present in the 19F NMR at δ −223.91 ppm (Fig. 2); the two lines coalesce into a singlet in the proton decoupled spectrum.
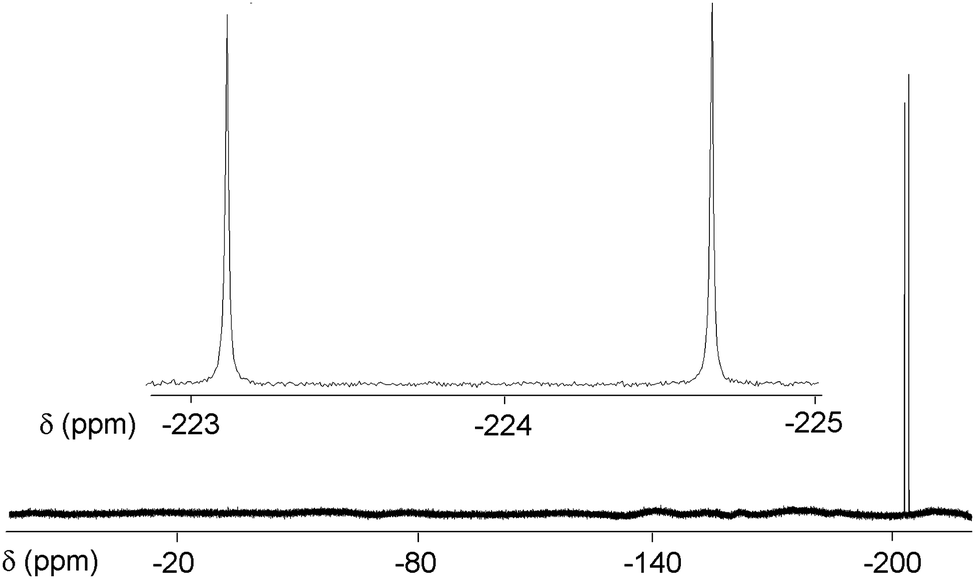 | ||
| Fig. 2 19F NMR (376 MHz, CDCl3) spectrum of compound HF@1. The δ scale is referenced to CFCl3. | ||
Solid-state NMR has been used for the characterization of molecular endofullerenes.25,27,28 The benefits of solid-state NMR include a much larger temperature range, even descending into the cryogenic regime5 and also the preservation of anisotropic interactions, which are averaged out in solution state NMR, but which contain information on the local structure, dynamics and symmetry. In the case of HF@1, the relevant anisotropic nuclear interactions include the chemical shift anisotropy (CSA), which reflects the electronic environment, and the dipole–dipole interaction between the 1H and 19F nuclei of the endohedral molecule.
Magic angle spinning (MAS) may be used to partly average out anisotropic interactions and produce high resolution spectra,29 which may still reveal the presence of significant anisotropies through spinning sidebands, which appear at multiples of the spinning frequency, and whose intensities are characteristic of the anisotropic interaction parameters.
The 1H solid-state MAS spectrum (Fig. 3), collected in a magnetic field of 19.96 T (850 MHz for 1H) at a spinning frequency of 20 kHz, displays an intense signal arising from the numerous exohedral protons and narrow peaks from the endohedral HF, reflecting the relatively isolated magnetic environment of the endohedral molecule and its rapid molecular motion inside the cage. This is in agreement with studies on other open-cage endofullerenes.28
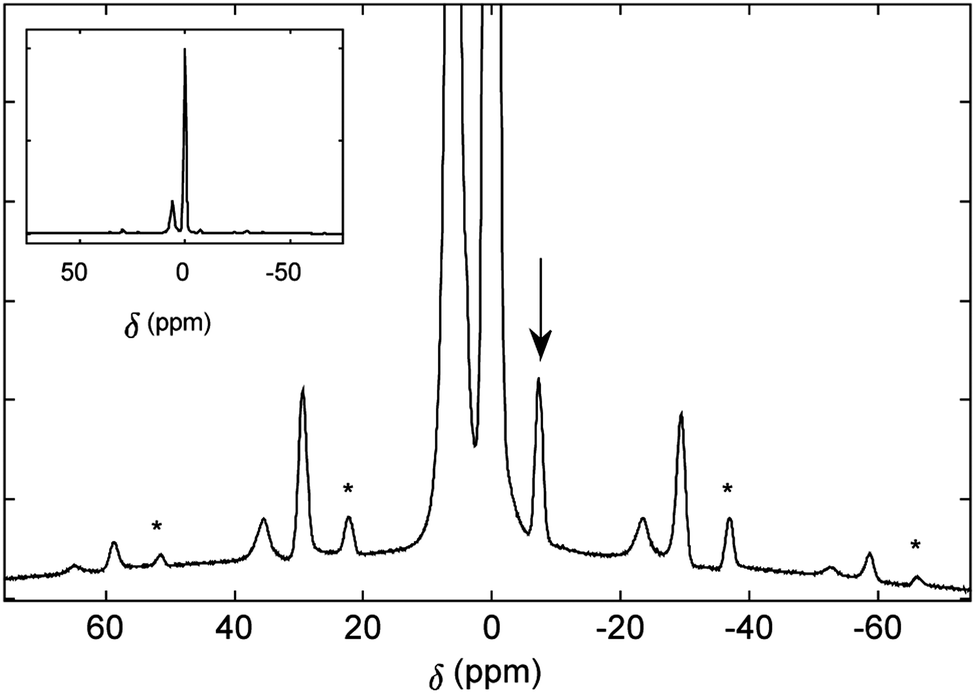 | ||
| Fig. 3 1H NMR spectrum of HF@1 recorded at 850 MHz (19.96 T) and a temperature of 263 K. The spinning frequency was 20 kHz. The −7 ppm centre band of the HF resonance is shown by the arrow. The spinning side bands are marked with asterisks. | ||
The 1H chemical shift is −7 ppm in the solid-state NMR spectrum, which is similar to the liquid-state value. The JHF coupling is not resolved in the solid-state NMR spectrum. Spinning sidebands can be seen at multiples of the spinning frequency, 20 kHz.
Magic-angle-spinning 19F spectra of HF@1 are shown in Fig. 4 for a set of spinning speeds. The 19F isotropic chemical shift is observed at −230 ppm.
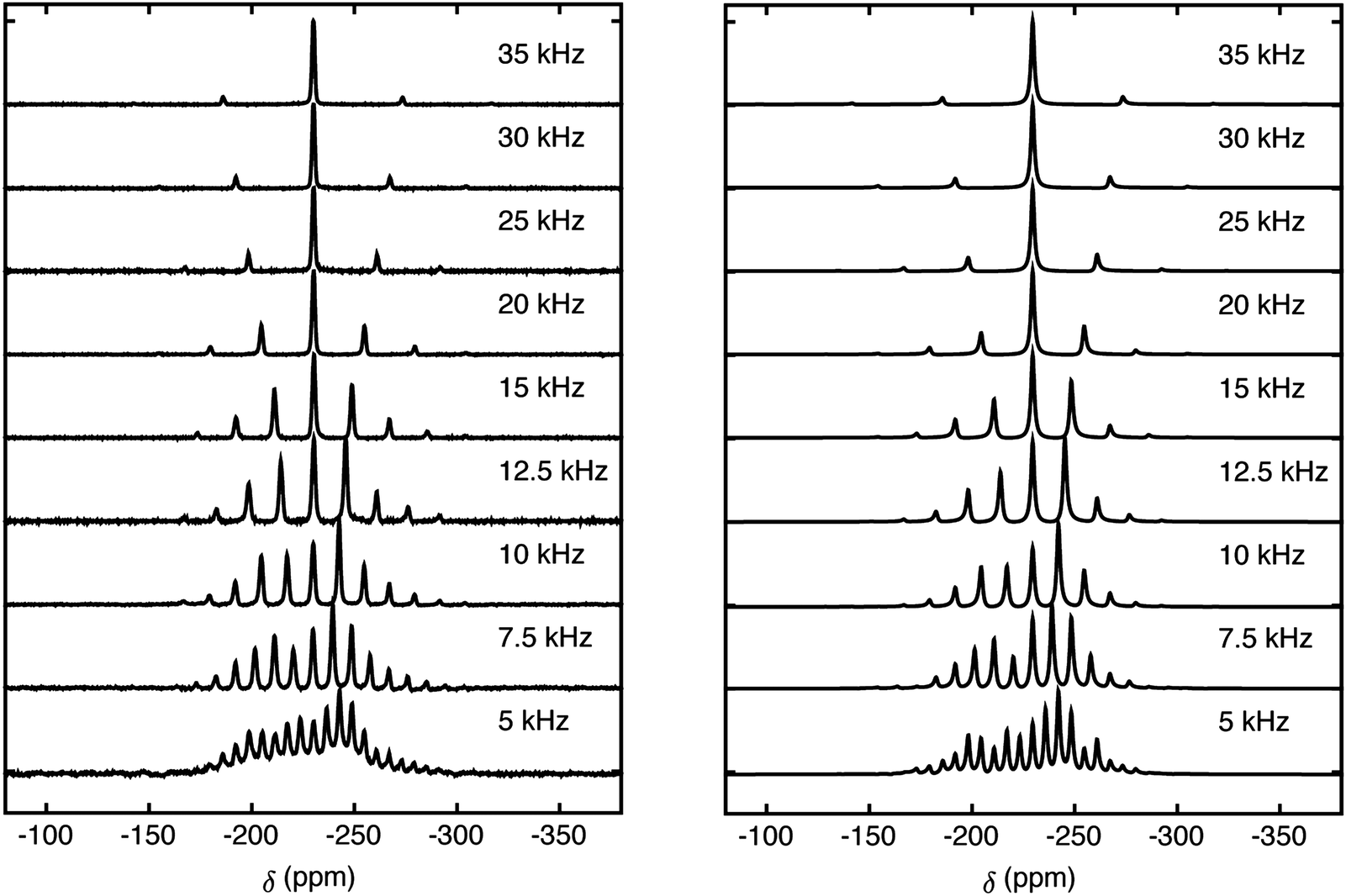 | ||
| Fig. 4 (left) 19F spectra of HF@1 recorded at 19.96 T and a temperature of 263 K, at the indicated spinning frequencies. The 19F chemical shift scale is referenced to β-polyvinylidene fluoride (PVDF); (right) simulated spectra with parameters determined from best fit to experiment. We used SPINACH30 and SIMPSON31 for simulations. Details of simulations can be found in ESI.† | ||
There are two anisotropic interactions that contribute to the generation of sidebands at low spinning frequencies, namely the dipole–dipole 1H–19F interaction and the 19F CSA. Numerical simulations of the NMR spectra were generated for combinations of isotropic chemical shift, asymmetry parameter, chemical shift anisotropy and the dipolar interaction using both SPINACH30 and SIMPSON.31 The direct dipolar interaction was determined to be −7.5 ± 2.5 kHz and the 19F chemical shift anisotropy as 44.1 ppm ± 1.7 ppm, with the biaxiality determined to be 0.6 ± 0.05. The 19F CSA tensor is found to be orientated at (α, β, γ) = (0, 90°, 0) with respect to the dipolar coupling tensor.
The direct dipolar coupling constant for an immobile 1H–19F pair separated by 0.91 Å would be −150 kHz. The small value of the observed dipolar coupling therefore indicates that the HF molecule rotates rapidly and almost isotropically inside the open fullerene cage. The second-rank order parameter of the HF molecule, indicating the degree of anisotropy of its rotational motion, is only ∼5%.
In this article we report the first HF-endofullerene and its study by solution and solid-state NMR. The solution NMR data shows a large 1H–19F J-coupling of 508 Hz, similar to that obtained for HF in the gas phase. The solid-state NMR spectra indicate that the HF molecule rotates rapidly and almost isotropically in the supramolecular complex. Unfortunately attempts to close the cage to give HF@C60 as reported for H2O@C6010 resulted in complete loss of HF.
This research was supported by the EPSRC (EP/I029451/1), the European Regional Development Fund (ERDF) Interreg-IVB, MEET project and the ERC. MC acknowledges the Royal Society for her University Research Fellowship. We are grateful for EPSRC Core Capability Funding (EP/K039466/1). We acknowledge the use of the IRIDIS High Performance Computing Facility and associated support services at the University of Southampton. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC, as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the ERDF. We would like to thank Dinu Iuga for experimental support at the 850 MHz NMR facility and Ilya Kuprov for help with SPINACH.
Notes and references
- M. H. Levitt and A. J. Horsewill, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 20130124 CrossRef PubMed.
- (a) L. Gan, D. Yang, Q. Zhang and H. Huang, Adv. Mater., 2010, 22, 1498 CrossRef CAS PubMed; (b) G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2010, 39, 817 RSC.
- S. Iwamatsu and S. Murata, Synlett, 2005, 2117 CrossRef CAS PubMed.
- M. H. Levitt, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 20120429 CrossRef PubMed.
- (a) N. J. Turro, A. A. Marti, J. Y. C. Chen, S. Jockusch, R. G. Lawler, M. Ruzzi, E. Sartori, S. C. Chuang, K. Komatsu and Y. Murata, J. Am. Chem. Soc., 2008, 130, 10506 CrossRef CAS PubMed; (b) Y. Li, X. Lei, S. Jockusch, J. Y. C. Chen, M. Frunzi, J. A. Johnson, R. G. Lawler, Y. Murata, M. Murata, K. Komatsu and N. J. Turro, J. Am. Chem. Soc., 2010, 132, 4042 CrossRef CAS PubMed; (c) C. Beduz, M. Carravetta, J. Y.-C. Chen, M. Concistré, M. Denning, M. Frunzi, A. J. Horsewill, O. G. Johannessen, R. Lawler, X. Lei, M. H. Levitt, Y. Li, S. Mamone, Y. Murata, U. Nagel, T. Nishida, J. Ollivier, S. Rols, T. Rõõm, R. Sarkar, N. J. Turro and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12894 CrossRef CAS PubMed; (d) S. Mamone, M. Concistre, E. Carignani, B. Meier, A. Krachmalnicoff, O. G. Johannessen, X. Lei, Y. Li, M. Denning, M. Carravetta, K. Goh, A. J. Horsewill, R. J. Whitby and M. H. Levitt, J. Chem. Phys., 2014, 140, 194306 CrossRef PubMed.
- Y. Rubin, T. Jarrosson, G.-W. Wang, M. D. Bartberger, K. N. Houk, G. Schick, M. Saunders and R. J. Cross, Angew. Chem., Int. Ed., 2001, 40, 1543 CrossRef CAS.
- K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238 CrossRef CAS PubMed.
- S. Iwamatsu, S. Murata, Y. Andoh, M. Minoura, K. Kobayashi, N. Mizorogi and S. Nagase, J. Org. Chem., 2005, 70, 4820 CrossRef CAS PubMed.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, Org. Lett., 2014, 16, 2970 CrossRef CAS PubMed.
- A. Krachmalnicoff, M. H. Levitt and R. J. Whitby, Chem. Commun., 2014, 50, 13037 RSC.
- S. Iwamatsu, T. Uozaki, K. Kobayashi, S. Re, S. Nagase and S. Murata, J. Am. Chem. Soc., 2004, 126, 2668 CrossRef CAS PubMed.
- Z. Xiao, J. Yao, D. Yang, F. Wang, S. Huang, L. Gan, Z. Jia, Z. Jiang, X. Yang, B. Zheng, G. Yuan, S. Zhang and Z. Wang, J. Am. Chem. Soc., 2007, 129, 16149 CrossRef CAS PubMed.
- Q. Zhang, T. Pankewitz, S. Liu, W. Klopper and L. Gan, Angew. Chem., Int. Ed., 2010, 49, 9935 CrossRef CAS PubMed.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613 CrossRef CAS PubMed.
- T. Futagoishi, M. Murata, A. Wakamiya, T. Sasamori and Y. Murata, Org. Lett., 2013, 15, 2750 CrossRef CAS PubMed.
- C. M. Stanisky, R. J. Cross and M. Saunders, J. Am. Chem. Soc., 2009, 131, 3392 CrossRef CAS PubMed.
- S. Iwamatsu, C. M. Stanisky, R. J. Cross, M. Saunders, N. Mizorogi, S. Nagase and S. Murata, Angew. Chem., Int. Ed., 2006, 45, 5337 CrossRef CAS PubMed.
- K. E. Whitener Jr., M. Frunzi, S. Iwamatsu, S. Murata, R. J. Cross and M. Saunders, J. Am. Chem. Soc., 2008, 130, 13996 CrossRef PubMed.
- K. E. Whitener Jr., R. J. Cross, M. Saunders, S. Iwamatsu, S. Murata, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2009, 131, 6338 CrossRef PubMed.
- D. Feller and K. A. Peterson, THEOCHEM, 1997, 400, 69 CrossRef CAS.
- (a) R. L. Jarry and W. Davis, J. Phys. Chem., 1953, 57, 600 CrossRef CAS; (b) D. F. Smith, J. Chem. Phys., 1958, 28, 1040 CrossRef CAS PubMed; (c) R. A. Oriani and C. P. Smyth, J. Am. Chem. Soc., 1948, 70, 125 CrossRef CAS.
- J. Cioslowski and A. Nanayakkara, Phys. Rev. Lett., 1992, 69, 2871 CrossRef CAS.
- DFT calculations were carried out using MO62-X/cc-pVDZ at B3LYP/6-31G(d) geometries. Details in the ESI†.
- (a) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872 CrossRef CAS; (b) G. A. Olah, T. Mathew, A. Goeppert, B. Torok, I. Bucsi, X. Y. Li, Q. Wang, E. R. Marinez, P. Batamack, R. Aniszfeld and G. K. Prakash, J. Am. Chem. Soc., 2005, 127, 5964 CrossRef CAS PubMed.
- M. Carravetta, A. Danquigny, S. Mamone, F. Cuda, O. G. Johannessen, I. Heinmaa, K. Panesar, R. Stern, M. C. Grossel, A. J. Horsewill, A. Samoson, M. Murata, Y. Murata, K. Komatsu and M. H. Levitt, Phys. Chem. Chem. Phys., 2007, 9, 4879 RSC.
- (a) C. MacLean and E. L. Mackor, J. Chem. Phys., 1961, 34, 2207 CrossRef CAS PubMed; (b) J. S. Muenter, J. Chem. Phys., 1970, 52, 6033 CrossRef PubMed; (c) J. S. Martin and F. Y. Fujiwara, J. Am. Chem. Soc., 1974, 96, 7632 CrossRef CAS.
- M. Concistrè, S. Mamone, M. Denning, G. Pileio, X. Lei, Y. Li, M. Carravetta, N. J. Turro and M. H. Levitt, Philos. Trans. R. Soc. London, Ser. A, 2013, 371, 20120102 CrossRef PubMed.
- M. Carravetta, Y. Murata, M. Murata, I. Heinmaa, R. Stern, A. Tontcheva, A. Samoson, Y. Rubin, K. Komatsu and M. H. Levitt, J. Am. Chem. Soc., 2004, 126, 4092 CrossRef CAS PubMed.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, Wiley, 2nd edn, 2008 Search PubMed.
- (a) H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. J. Hore and I. Kuprov, J. Magn. Reson., 2011, 208, 179 CrossRef CAS PubMed; (b) L. J. Edwards, D. V. Savostyanov, A. A. Nevzorov, M. Concistrè, G. Pileio and I. Kuprov, J. Magn. Reson., 2013, 235, 121 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data and details of calculations. See DOI: 10.1039/c5cc00499c |
| This journal is © The Royal Society of Chemistry 2015 |