
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceenantio-Enriched CPL-active helicene–bipyridine–rhenium complexes†
Nidal
Saleh
a,
Monika
Srebro
b,
Thibault
Reynaldo
a,
Nicolas
Vanthuyne
c,
Loïc
Toupet
a,
Victoria Y.
Chang
d,
Gilles
Muller
d,
J. A. Gareth
Williams
e,
Christian
Roussel
c,
Jochen
Autschbach
*f and
Jeanne
Crassous
*a
aInstitut des Sciences Chimiques de Rennes, UMR 6226, Institut de Physique de Rennes, UMR 6251, Campus de Beaulieu, CNRS-Université de Rennes 1, 35042 Rennes Cedex, France. E-mail: jeanne.crassous@univ-rennes1.fr
bFaculty of Chemistry, Jagiellonian University, 30-060 Krakow, Poland
cAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France
dDepartment of Chemistry, San José State University, San José, CA 95192-0101, USA
eDepartment of Chemistry, University of Durham, Durham, DH1 3LE, UK
fDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
First published on 23rd January 2015
Abstract
The incorporation of a rhenium atom within an extended helical π-conjugated bi-pyridine system impacts the chiroptical and photophysical properties of the resulting neutral or cationic complexes, leading to the first examples of rhenium-based phosphors that exhibit circularly polarized luminescence.
2,2′-Bipyridine (bipy) derivatives are widely used N,N′-bidentate ligands in coordination chemistry, giving access to a great variety of complexes.1 The luminescence properties of d6 transition metal polypyridyl complexes have been increasingly studied for the development of new metal-based luminescent materials and sensing probes.2 Among them, [Re(N,N′)(CO)3X]0/+ complexes (X = halide, pyridyl (py) or isocyanide (CNR)) exhibit room-temperature (RT) phosphorescence from triplet metal-to-ligand (ML) and/or intraligand charge-transfer (ILCT) states.3,4 Such d6-complexes find applications as electroswitchable emissive systems,5a cellular imaging agents,5b,c chromophores for photoredox chemistry,5detc. It would therefore be of great interest to develop chiral analogues6 in order to benefit from the chiral version of emission, namely circularly polarized luminescence (CPL) which may potentially be used in cryptography or for 3D-displays.7,8
In this communication, we describe the synthesis of tricarbonyl ReI complexes of general formula [Re(N∧N′)(CO)3X]0/+ (X = halide, pyridyl or isocyanide) with N∧N′ being either achiral 3-(2-pyridyl)-4-aza[4]-helicene (1a) or chiral 3-(2-pyridyl)-4-aza[6]-helicene (M- and P-1b) (Scheme 1). The stereochemical features of these novel d6-complexes are presented in detail. The chiroptical properties of enantio-enriched samples and the non-polarized and circularly polarized phosphorescence were measured experimentally and analyzed using quantum-chemical calculations.
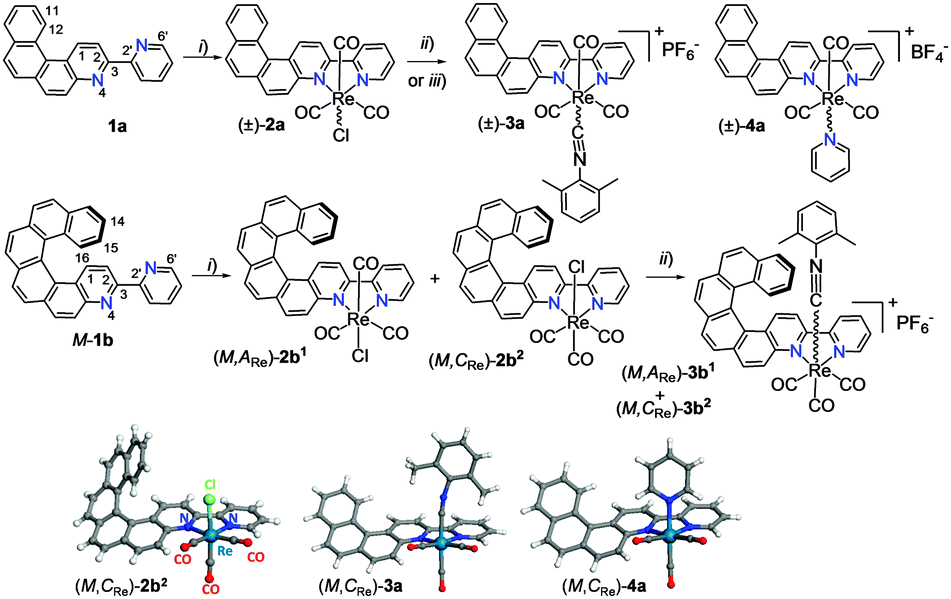 | ||
| Scheme 1 Synthesis of rhenium complexes 2a–4a and enantio-enriched (M,ARe)-2b1, (M,CRe)-2b2 and (M,ARe*)-3b1,2 from respectively [4]helicene-bipy, 1a and M-[6]helicene-bipy, M-1b. (i) Re(CO)5Cl, toluene, reflux; (ii) AgOTf, EtOH/THF, then 2,6-dimethylphenyl isocyanide, THF, NH4PF6, 75–80%; (iii) AgBF4, CH3CN, reflux then pyridine, THF, 80%. X-ray structures of racemic 2b2, 3a and 4a (only (M,CRe) stereoisomers are shown).10 | ||
ReI-complex 2a was obtained in 85% yield as a yellow-orange precipitate upon refluxing a solution of 1a8g and Re(CO)5Cl in toluene for 5 hours (Scheme 1). It was fully characterized by multinuclear NMR spectroscopy (one set of peaks), by elemental analysis, UV-vis and emission spectroscopies. As compared to ligand 1a, the 1H NMR spectrum of 2a shows strongly deshielded signals (except for H2, H12 and H6′) with Δδ up to +0.8 ppm for H5 (see ESI†). The UV-vis spectrum of ligand 1a in CH2Cl2 displays a strong band at 295 nm (ε > 50 × 103 M−1 cm−1), accompanied by several structured bands of lower intensity between 300 and 400 nm. Meanwhile, complex 2a shows several absorption bands between 230 and 370 nm (ε ∼ 30–43 × 103 M−1 cm−1) that can be assigned to intraligand π–π* transitions and a broad, low-energy absorption band between 370 and 480 nm (λmax = 398 nm, ε = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1) related to the incorporation of the ReI metal and predominantly assigned as ILCT with contributions of MLCT character (vide infra). The absorption maximum at 398 nm appears red-shifted compared to the corresponding band in Re(2,2′-bpy)(CO)3Cl3f (350 nm) indicating extended π-conjugation.
700 M−1 cm−1) related to the incorporation of the ReI metal and predominantly assigned as ILCT with contributions of MLCT character (vide infra). The absorption maximum at 398 nm appears red-shifted compared to the corresponding band in Re(2,2′-bpy)(CO)3Cl3f (350 nm) indicating extended π-conjugation.
ReI complex 2a is red-phosphorescent in CH2Cl2 at RT (λphosmax = 678 nm, ϕ = 0.11%, τ = 25 ns, see ESI†). The phosphorescence originates from the triplet charge-transfer state. It is facilitated by spin–orbit coupling at the rhenium heavy atom and bathochromically shifted compared to that of Re(2,2′-bpy)(CO)3Cl (λphosmax = 610 nm).3f At 77 K, the phosphorescence of 2a is significantly shifted to shorter wavelengths (λphosmax = 550 nm, τ = 7.9 μs). Such a hypsochromic shift is usually explained by inversion of the energies of 3π–π* and 3MLCT triplet states and/or by rigidification of the system.3 Note that as usual in this class of complexes, the quantum yield at RT is rather low.3 In comparison, charged complexes of formula [Re(N∧N′)(CO)3py]+ or [Re(N∧N′)(CO)3CNR]+ typically display superior luminescence efficiency due to a stronger ligand field. For this reason, complexes 3a and 4a were prepared in good yields from 1a, according to Scheme 1. They were fully characterized by multinuclear NMR spectroscopy, elemental analysis, UV-vis spectroscopy, emission and X-ray crystallography. The 3a and 4a compounds crystallize in Fdd2 and P21/n centrosymmetric space groups respectively (Scheme 1). At this stage, it is worth noting that complexes 2a–4a are chiral at the rhenium centre,‡9 since the Re atoms adopt a slightly distorted octahedral geometry, with three carbonyl groups being fac-oriented around the ReI, as classically seen in such rhenium(I) tricarbonyl diimine complexes.3 The equatorial planes are defined by the chelate bipyridine ligand and two trans carbonyls. A third carbonyl and either the chlorine, the isocyanide or the pyridine are placed in the apical positions. Note that in structures 3a,4a the [4]helicene-bpy ligand exhibits a helicity angle (defined as angle between the terminal rings of the helicene moiety) of ∼35° and the cyanide and pyridine ligand are directed towards it, thus defining the (P,ARe) and (M,CRe) stereochemistry.10 However, in solution, the helicene is not configurationally stable, and the Re center readily epimerizes (vide infra). As expected, the charged complexes displayed improved photophysical properties with similar UV-visible and emission spectra as for 2a (see ESI†), but with higher quantum yields (3a: 16%; 4a: 8.3%). These results prompted us to prepare tricarbonylrhenium(I) complexes bearing a configurationally stable enantiopure [6]helicene-bipy ligand.
Racemic 1b was reacted with Re(CO)5Cl in refluxing toluene for 5 hours, yielding after purification by column chromatography two distinct diastereomeric Re(I) complexes (2b1 and 2b2, with 28% and 52% yields, respectively) as evidenced by 1H and 13C NMR spectroscopy (for example H15: 6.7 ppm for 2b1 and 6.9 ppm for 2b2, see ESI†). Complex 2b2 crystallizes in a centrosymmetric space group (P21/C) in which two enantiomeric structures, namely (M,CRe)- and (P,ARe)-2b2 are present (Scheme 1).10 Note that a substantial distortion results from the bite angles between the chelating N atoms of the helicenic ligand, the rhenium centre and the chloride ligand ranging between 82.6 and 84.3°. In complex 2b2 the chlorine atom is directed towards the helicene moiety, whereas it directs outwards from the helicene core in the enantiomeric complexes (M,ARe) and (P,CRe)-2b1. The helicity of the aza[6]helicene moiety ranges between 47.0–66.2°, which is typical for aza[6]helicene derivatives (58° for carbo[6]helicene).8 Finally, complexation with Re affords an extended π-conjugation over the whole molecule, as evidenced by the small NCCN dihedral angles between the two chelating pyridine moieties (−3.1–6.0°). The extended π-conjugation and the metal-ligand interaction are evidenced by UV-vis spectroscopy since 2b1,2 display similar UV-vis spectra with a set of several bands between 330 and 450 nm (ε ∼ 7–25 × 103 M−1 cm−1) that are bathochromically shifted and more intense compared to ligand 1b, together with a very weak band observed between 450 and 500 nm (see Fig. S21, ESI†). Calculations at the BHLYP/SV(P) level with a continuum solvent model for CH2Cl2 reproduce well these data and show that the low-energy band of the spectrum is dominated by an ILCT transition, π(helicene) → π*(N∧N′), while the medium-energy bands are mostly π-to-π* ‘CT-like’ transitions localized within the helicene moiety (vide infra, ESI†) in agreement with assignments of absorption spectra of related rhenium(I) systems, in particular for complexes with large π-conjugated ligands.4c–e The overall contribution of the Re orbitals is low, meaning that the primary effect of the metal is to rigidify the system and induce strong charge transfer from the helical π-system to the bipy N∧N′ part of the ligand. The simulated spectral shapes and band positions agree well with experiment. It is possible, though, that the overall involvement of Re orbitals in the absorption transitions is somewhat underestimated by the BHLYP functional (vide infra). ReI complexes 2b1,2 are red-phosphorescent emitters in CH2Cl2 at RT (2b1: λphosmax = 680 nm, ϕ = 0.13%, τ = 27 ns; 2b2: λphosmax = 673 nm, ϕ = 0.16%, τ = 33 ns; for details see ESI†). At 77 K, these complexes display phosphorescence at shorter wavelengths (2b1: λphosmax = 560 nm, τ = 46 μs; 2b2: λphosmax = 554 nm, τ = 43 μs) (vide supra). Note that the emission properties of diastereomers 2b1,2 are only slightly different and (for τ and ϕ) within the uncertainty in the measurements (see ESI†).
Enantiopure complexes (M,ARe)-2b1 and (M,CRe)-2b2 were then prepared from enantiopure M-1b (their mirror-images (P,CRe)-2b1 and (P,ARe)-2b2 from P-1b). Enantiopure complexes 2b1,2 display similar molar rotation (MR) values to 1b in CH2Cl2 {(P,CRe)-(+)-2b1: [ϕ]23D = +9260 degree cm2 dmol−1 (±5%), calc. BHLYP +12721; (P,ARe)-(+)-2b2: [ϕ]23D = +10260 (±5%), calc. BHLYP +11888; P-(+)-1b:8g [ϕ]23D = +12000 (±5%), calc. BHLYP +14176, see ESI†}. The ECD spectrum of P-2b1 shows a strong negative band around 261 nm (Δε = −114 M−1 cm−1) and strong positive ones at 350 (+81 M−1 cm−1) and 368 nm (+76 M−1 cm−1) accompanied by weaker bands between 380 and 450 nm (20–40 M−1 cm−1) and an even weaker one around 480 nm but of opposite sign (Δε ∼ −0.6 M−1 cm−1). Diastereomeric complex (P,ARe)-(+)-2b2 exhibits the same ECD active bands as 2b1 but they are more intense. A comparison with experimental ECD of 1b enantiomers is displayed in Fig. 1. The calculated spectra of 2b1,2 (BHLYP/SV(P) with a continuum solvent model for CH2Cl2) qualitatively agree well with the experimental results (Fig. 3 and Fig. S5, ESI†). A detailed analysis of dominant excitations in the low- and medium-energy parts of the simulated spectra of 2b1,2 indicates that the low-energy tail of the first positive ECD band is caused by excitation no. 1 calculated at E = 3.3 eV (375 nm). The excitation can be assigned as a π–π* ILCT transition involving the helicene-centered HOMO (H), H − 1, and the bipyridine N∧N′-centered LUMO (L), for example for 2b1: H–L 51% and H − 1–L 18% (see Fig. 3 and ESI†). The next dominant 2b1,2 excitation is no. 5 calculated at E = 3.8 eV (330 nm) with the strongest rotatory strength. It involves two main contributions from π and π* orbital pairs localized mostly in the helicene moiety: H–L + 1 and H − 1–L + 1 (respectively 43% and 25% for 2b1). The excitation reveals partial CT character.
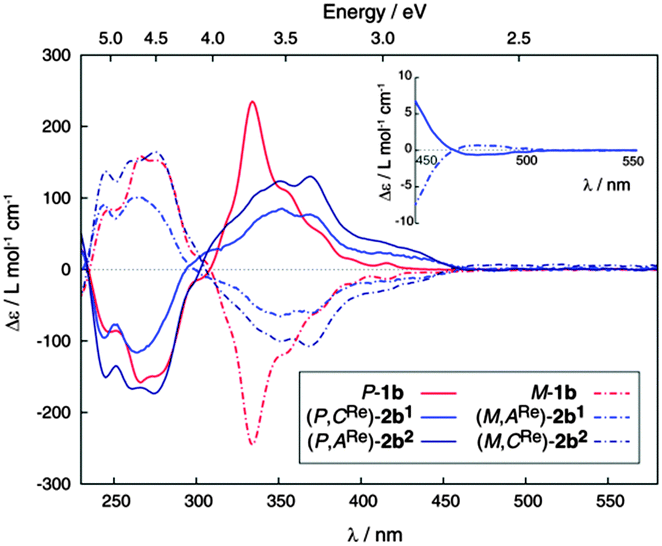 | ||
| Fig. 1 Experimental CD spectra of enantiopure M-(−)-(dotted red) and P-(+)-1b (plain red) and enantiopure ReI complexes (M,ARe)-(−)-2b1 and (P,CRe)-(+)-2b1 (light blue) and (M,CRe)-(−)-2b2 and (P,ARe)-(+)-2b2 (dark blue). Inset: CD spectra of (−)- and (+)-2b1 between 450–550 nm. | ||
A novel aspect of these rhenium(I) helicene-based complexes is that they are CPL active (Fig. 2, top panels).7,8 To the best of our knowledge, these are the first examples of CPL-active phosphorescent rhenium complexes. Indeed phosphorescent (P,ARe) and (M,CRe)-2b2 enantiomers displayed mirror-imaged CPL spectra (Fig. 2) with opposite glum values ((P,ARe)-2b2: +3.1 × 10−3 and (M,CRe)-2b2: −2.8 × 10−3) around the emission maximum (∼670 nm). These values are of the same order as for the 1b ligand enantiomers (glum∼ ±10−3) but lower than those of previously published platina[6]helicenes (glum ∼ ±10−2),8e because Re orbitals are less involved in the helical π-system of the molecule (vide supra).
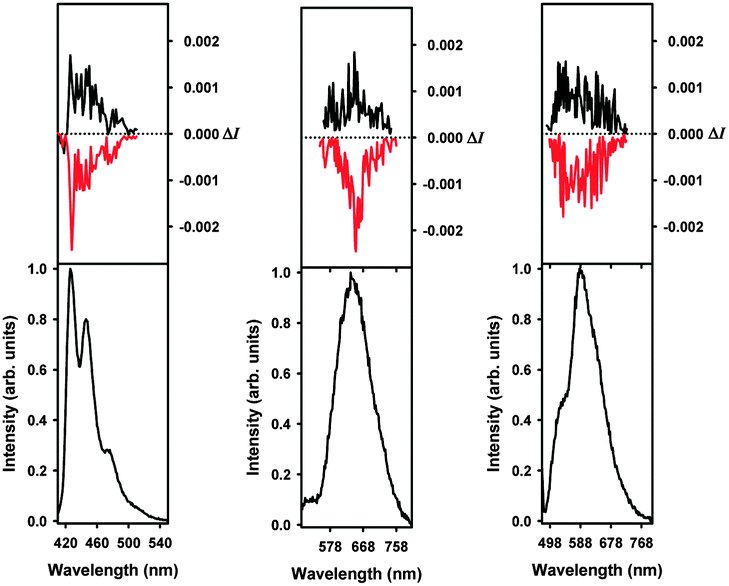 | ||
| Fig. 2 CPL (upper curves within each panel) and total luminescence (lower curves within each panel) spectra of M-(−)-1b (left red), P-(+)-1b (left black), M-(−)-2b2 (middle red), P-(+)-2b2 (middle black), M-(−)-3b1,2 (right red), P-(+)-3b1,2 (right black) in degassed CH2Cl2 solution (1 mM) at 295 K, upon excitation at 400, 456–461, and 458–468 nm, respectively. | ||
In order to improve the efficiency of the chiroptical and photophysical properties, tricarbonyl-isocyanide-helicene-bipy-ReI complex M-3b was prepared (see Scheme 1) in 75% yield from either (M,ARe)-(−)-2b1 or (M,CRe)-(−)-2b2. In this complex, the Re center appeared labile and 3b was obtained as a mixture of (M,ARe)-3b1 and (M,CRe)-3b2 as observed by 1H and 13C NMR spectroscopy (diastereomeric ratio 50:50, see Fig. S27, ESI†) regardless of the diastereomeric purity of the starting compound used (either 2b1 or 2b2 or 2b1,2). Nevertheless, as expected, this diastereomeric mixture exhibited an improved quantum yield (λphosmax = 598 nm, ϕ = 6%, τ = 79 μs; see ESI†) as compared to 2b1,2. The UV-vis spectrum of 3b1,2 displays the same shape as 2b1 (see Fig. S21, ESI†). Compared to (P,CRe)-2b1 and (P,ARe)-2b2, cationic diastereomeric mixture of ReI complexes P-3b1,2 demonstrates an additional positive CD-active band around 450 nm (Δε = 17.5 M−1 cm−1). As for 2b1,2, this latter band does not involve the Re center, but corresponds to the H–L transition (>74%) with strong charge transfer from the π-helicene to the bipy moiety, as evidenced by BHLYP calculations (see Fig. 3 and ESI†). The appearance of the 450 nm band is caused mainly by a bathochromic shift of the first singlet excitation. This charge-transfer excitation is likely responsible for the enhancement of molar rotations as compared to 2b1,2 {(P,ACRe)-(+)-3b: [ϕ]23D = 15040 degree cm2 dmol−1 (±5%) (C = 8.8 × 10−5 M, CH2Cl2); (M,ACRe)-(−)-3b: [ϕ]23D = −14230 (±5%) (C = 9.7 × 10−5 M, CH2Cl2); calc. BHLYP Boltzmann average for 3b1,2 conformers is +14034 degree cm2 dmol−1 for the P-isomers, see ESI†}.
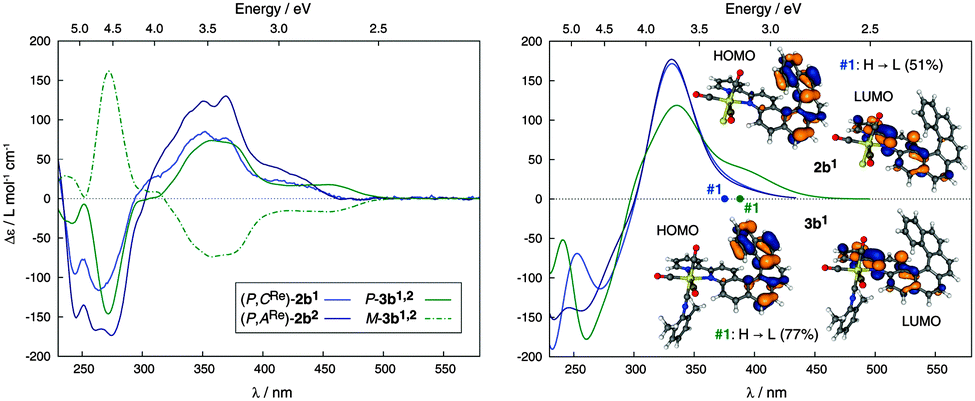 | ||
| Fig. 3 Left: experimental CD spectra of enantiopure complexes (P,CRe)-(+)-2b1 (light blue), (P,ARe)-(+)-2b2 (dark blue) and of (M,CARe)-(−)-3b1,2 (dotted green) and (P,CARe)-(+)-3b1,2 (plain green). Right: calculated CD spectra of (P,CRe)-2b1, and (P,ARe)-2b2 and Boltzmann-averaged spectrum for P-3b1,2 conformers. No spectral shifts were applied. View of HOMO and LUMO of 2b1 and 3b1 (0.04 au). First excitation energies indicated by dots on the abscissa. | ||
Quantum-chemical calculations of luminescence properties have been performed for 2b1,2 and 3b1,2. The results support the experimental assignments: The energies of T1 → S0 phosphorescence transitions (∼2.1 eV) are similar for both 2b1,2 and 3b1,2 and agree fairly well with the experimental data (Table S5, ESI†). An overestimation of the calculated versus measured energies is consistent with a blue-shift of calculated 2b1,2 and 3b1,2 absorption and CD spectra. The emission energies from spin–orbit (SO) calculations agree with non-SO calculations but the former allow predictions of the phosphorescence lifetimes. Application of the BHLYP functional along with the Tamm-Dancoff approximation (see ESI†) resulted in much too high emission lifetimes (Table S6, ESI†). As the involvement of Re orbitals facilitates the formally spin-forbidden T1 → S0 phosphorescence transitions via spin–orbit coupling, decreasing the corresponding lifetimes, too high τ calculated with BHLYP may indicate that the metal orbital contributions to the frontier MOs are somewhat too small. The performance of a given functional for singlet vs. triplet transitions is not necessarily the same. When applying a computational protocol for emission lifetimes devised recently by Mori et al.11 for organometallic complexes (full TDDFT with the B3LYP functional), a dramatic improvement of the lifetimes and some lowering of the emission energies (to ∼1.9 eV) was obtained (Table S7, ESI†), which correlates with increased participation of Re orbitals in the frontier MOs at the triplet geometries. Notably the experimental trend of an increase in emission lifetime by roughly an order of magnitude when going from 2b1,2 to 3b1,2 is correctly reproduced with B3LYP and qualitatively consistent with lesser metal orbital participation (lesser MLCT character) in the T1 emission transitions for 3b1,2 as compared to 2b1,2 (see ESI†).§4a,b
Finally, mirror-imaged CPL spectra were obtained in CH2Cl2 for (M,ACRe)-3b1,2 and (P,ACRe)-3b1,2 (Fig. 2) with respective glum values of −0.0015 and +0.0013. Overall, cationic ReI complexes display similar CPL characteristics as neutral ones, but combined with a higher quantum yield, the polarized emitted light is stronger. Although the Re d orbitals are not strongly involved in the electronic π-systems of these novel metallo-helicenes, the metal helps to increase the π-conjugation pathway and promotes charge-transfer excitations within the π-helical ligand. In addition, the presence of the rhenium heavy atom makes these complexes chiral phosphors with unprecedented CPL activity.
We thank the Ministère de l′Education Nationale, de la Recherche et de la Technologie, the CNRS, the ANR (10-BLAN-724-1-NCPCHEM and 12-BS07-0004-METALHEL-01) and the LIA Rennes-Durham for financial support. J.A. acknowledges the UB Center for Computational Research and thanks the National Science Foundation (CHE 1265833) for financial support. M.S. thanks the Foundation for Polish Science Homing Plus program co-financed by the European Regional Development Fund and the Ministry of Science and Higher Education in Poland scholarship. G.M. thanks NIH MBRS (1 SC3 GM089589-05 and 3 S06 GM008192-27S1) and the Henry Dreyfus Teacher-Scholar Award for financial support.
Notes and references
- (a) G. Chelucci and R. P. Thummel, Chem. Rev., 2002, 102, 3129 CrossRef CAS PubMed; (b) H.-L. Kwong, H.-L. Yeung, C.-T. Yeung, W.-S. Lee, C.-S. Lee and W.-L. Wong, Coord. Chem. Rev., 2007, 251, 2188 CrossRef CAS PubMed.
- H. Le Bozec and V. Guerchais, Molecular Organometallic Materials for Optics, Topics in Organometallic Chemistry series, Springer, 2009 Search PubMed.
- (a) K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603 CrossRef CAS PubMed; (b) V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC; (c) C.-C. Ko, A. W.-Y. Cheung, L. T.-L. Lo, J. W.-K. Siu, C.-O. Ng and S.-M. Yiu, Coord. Chem. Rev., 2012, 256, 1546 CrossRef CAS PubMed; (d) M. Panigati, M. Mauro, D. Donghi, P. Mercandelli, P. Mussini, L. de Cola and G. D'Alfonso, Coord. Chem. Rev., 2012, 256, 1621 CrossRef CAS PubMed; (e) A. J. Lee, Chem. Rev., 1987, 87, 711 CrossRef; (f) M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998 CrossRef CAS.
- (a) M. K. Kuimova, W. Z. Alsindi, J. Dyer, D. C. Grills, O. S. Jina, P. Matousek, A. W. Parker, P. Portius, X. Z. Sun, M. Towrie, C. Wilson, J. Yang and M. W. George, Dalton Trans., 2003, 3996 RSC; (b) H. van der Salm, M. G. Fraser, R. Horvath, S. A. Cameron, J. E. Barnsley, X.-Z. Sun, M. W. George and K. C. Gordon, Inorg. Chem., 2014, 53, 3126 CrossRef CAS PubMed; (c) H.-J. Nie, X. Chen, C.-J. Yao, Y.-W. Zhong, G. R. Hutchison and J. Yao, Chem. – Eur. J., 2012, 18, 14497 CrossRef CAS PubMed; (d) T. Yu, D. P.-K. Tsang, V. K.-M. Au, W. H. Lam, M.-Y. Chan and V. W.-W. Yam, Chem. – Eur. J., 2013, 19, 13418 CrossRef CAS PubMed; (e) R. Horvath, M. G. Fraser, S. A. Cameron, A. G. Blackman, P. Wagner, D. L. Officer and K. C. Gordon, Inorg. Chem., 2013, 52, 1304 CrossRef CAS PubMed.
- Selected: (a) K. M. C. Wong, S. C. F. Lam, C. C. Ko, N. Y. Zhu, V. W. W. Yam, S. Roue, C. Lapinte, S. Fathallah, K. Costuas, S. Kahlal and J.-F. Halet, Inorg. Chem., 2003, 42, 7086 CrossRef CAS PubMed; (b) A. W.-T. Choi, V. M.-W. Yim, H.-W. Liu and K. K.-W. Lo, Chem. – Eur. J., 2014, 20, 9633 CrossRef CAS PubMed; (c) S. Clède, F. Lambert, C. Sandt, Z. Gueroui, M. Réfrégiers, M.-A. Plamont, P. Dumas, A. Vessières and C. Policar, Chem. Commun., 2012, 48, 7729 RSC; (d) H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544 CrossRef CAS PubMed.
- (a) Y. H. Zhou, J. Li, T. Wu, X. P. Zhao, Q. L. Xu, X. L. Li, M. B. Yu, L. L. Wang, P. Sun and Y. X. Zheng, Inorg. Chem. Commun., 2013, 29, 18 CrossRef CAS PubMed; (b) M. Q. Sans and P. Belser, Coord. Chem. Rev., 2002, 229, 59 CrossRef CAS.
- H. Maeda and Y. Bando, Pure Appl. Chem., 2013, 85, 1967 CrossRef CAS.
- Selected examples of CPL active helicenes: (a) J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808 CrossRef CAS PubMed; (b) Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080 CrossRef CAS PubMed; (c) K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899 CrossRef CAS PubMed; (d) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684 CrossRef CAS PubMed; (e) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson Jr., G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915 RSC; (f) K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555 CrossRef CAS PubMed; (g) N. Saleh, B. Moore, II, M. Srebro, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, K. K. Deol, G. Muller, J. Autschbach and J. Crassous, Chem. – Eur. J., 2015, 21, 1673 CrossRef CAS PubMed.
- For enantiopure “chiral at rhenium” complexes see: (a) S. J. Lee and W. Lin, J. Am. Chem. Soc., 2002, 124, 4554 CrossRef CAS PubMed; (b) F. Bock, F. Fischer and W. A. Schenk, J. Am. Chem. Soc., 2006, 128, 68 CrossRef CAS PubMed; (c) J.-D. Chen and F. A. Cotton, J. Am. Chem. Soc., 1991, 113, 2509 CrossRef CAS; (d) J. H. Merrifield, C. E. Strouse and J. A. Gladysz, Organometallics, 1982, 1, 1204 CrossRef CAS; (e) W. E. Buhro, A. Wong, J. H. Merrifield, G.-Y. Lin, A. C. Constable and J. A. Gladysz, Organometallics, 1983, 2, 1852 CrossRef CAS; (f) P. R. Lassen, L. Guy, I. Karame, T. Roisnel, N. Vanthuyne, C. Roussel, X. Cao, R. Lombardi, J. Crassous, T. B. Freedman and L. A. Nafie, Inorg. Chem., 2006, 45, 10230 CrossRef CAS PubMed; (g) F. De Montigny, L. Guy, G. Pilet, N. Vanthuyne, C. Roussel, R. Lombardi, T. B. Freedman, L. A. Nafie and J. Crassous, Chem. Commun., 2009, 4841 RSC; (h) J. W. Faller and A. R. Lavoie, Organometallics, 2000, 19, 3957 CrossRef CAS; (i) W. K. Rybak, A. Skarzynska and T. Glowiak, Angew. Chem., Int. Ed., 2003, 42, 1725 CrossRef CAS PubMed; (j) C. M. Alvarez, R. Carrillo, R. Garcia-Rodriguez and D. Miguel, Chem. Commun., 2011, 47, 12765 RSC; (k) E. Tazacs, A. Escande, N. Vanthuyne, C. Roussel, C. Lescop, E. Guinard, C. Latouche, A. Boucekkine, J. Crassous, R. Réau and M. Hissler, Chem. Commun., 2012, 48, 6705 RSC.
- For the stereochemical descriptors see ESI† and A. von Zelewsky, Stereochemistry of Coordination Compounds, J. Wiley & Sons, Chichester, 1996 Search PubMed.
- K. Mori, T. P. M. Goumans, E. van Lenthe and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14523 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic, spectroscopic and computational details. CCDC 857156, 939180 and 942143. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00453e |
| ‡ Note that enantiopure (+) and (−) complexes (1a)Re(CO)3Br were prepared by chiral HPLC and they displayed very weak CD activity (Δεmax = 20 M−1 cm−1, see Fig. S23, ESI†). |
| § The direct comparison between experimental and calculated lifetimes must be treated with some caution, as the former are also affected by non-radiative decay pathways, which may be non-negligible even at 77 K. We assume that the non-radiative decay rates for the complexes are similar under these conditions. |
| This journal is © The Royal Society of Chemistry 2015 |