
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative Heck desymmetrisation of 2,2-disubstituted cyclopentene-1,3-diones†
S. E.
Walker
,
C. J. C.
Lamb
,
N. A.
Beattie
,
P.
Nikodemiak
and
A.-L.
Lee
*
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: A.Lee@hw.ac.uk; Tel: +44 (0)131-4518030
First published on 3rd February 2015
Abstract
Oxidative Heck couplings have been successfully developed for 2,2-disubstituted cyclopentene-1,3-diones. The direct coupling onto the 2,2-disubstituted cyclopentene-1,3-dione core provides a novel expedient way of enantioselectively desymmetrising all-carbon quaternary centres.
The 2,2-disubstituted cyclopentene-1,3-dione core is found in several biologically active natural products, including madindolines A and B,1 similin A2 and ochroleucin A1,3 and metabolites such as preussidone4 and involutone5 (e.g.Fig. 1). As such, a direct, catalytic method for accessing such motifs would be of synthetic value, but no examples of such methods were available at the commencement of this project.6,7 We therefore aimed to develop a Heck-type8 desymmetrisation on easily accessible substrates 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 using chiral enantiopure ligands10 (Scheme 1),11 as this is in principle one of the most direct ways of obtaining the stereogenic all-carbon quaternary centre found in 2.12
9 using chiral enantiopure ligands10 (Scheme 1),11 as this is in principle one of the most direct ways of obtaining the stereogenic all-carbon quaternary centre found in 2.12
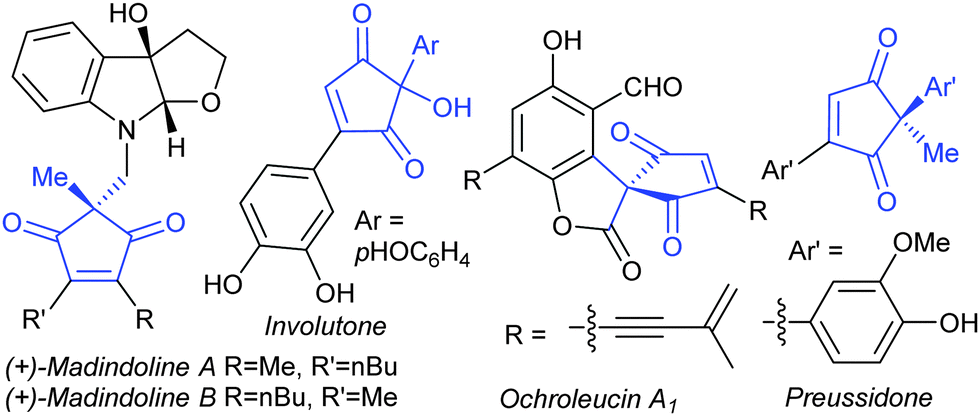 | ||
| Fig. 1 Examples of natural products containing 2,2-disubstituted cyclopentene-1,3-dione cores. | ||
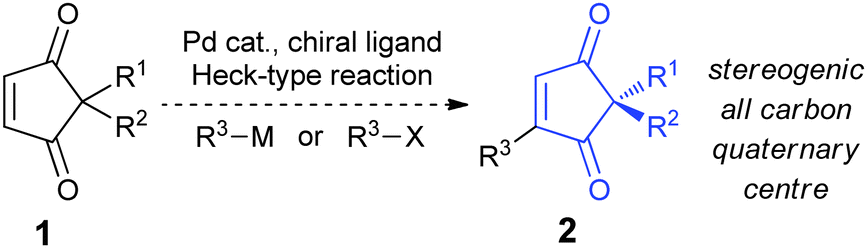 | ||
| Scheme 1 Heck-type desymmetrisation of 2,2-disubstituted cyclopentene-1,3-diones. | ||
During the preparation of this manuscript, an elegant base-mediated organocatalytic alkylation method was reported by Mukherjee and co-workers using nitroalkyls as the alkylating agent.13 However, this alternative approach is necessarily limited to alkylations (R3 = alkyl in 2), which precludes it as a method towards non-alkyl substituted target products such as involutone, ochroleucin A1 and preussidone (Fig. 1). Therefore, the development of a Heck-type desymmetrisation, capable of arylating enediones 1, is still highly relevant for the access of other 2,2-disubstituted cyclopentene-1,3-dione targets.
Despite their obvious potential, Heck-type reactions have not previously been reported on cyclopentene-1,3-dione substrates such as 1. This lack of literature precedence is most likely due to the fact that cyclic enones are notoriously reluctant to undergo Pd(0)-catalysed Mizoroki–Heck couplings and will often produce the conjugate addition products instead, as well as being stereochemically precluded from undergoing the final step in the traditional Pd(0) Heck cycle: the syn β-H elimination.14 As substrates 1 are expected to be challenging substrates for the Heck-type reaction, our initial aim was to develop a racemic Heck-type protocol for 1, followed by enantioselective desymmetrisations. Our successful efforts toward this goal are presented herein.
We decided to utilise Pd(II)-catalysed oxidative Heck10d,15,16 methods as they have recently been shown to be more compatible with cyclic enones than Pd(0)-catalysed Heck couplings.17 Nevertheless, examples of successful oxidative Heck couplings on cyclic enone derivatives are still fairly scarce18 and do not include any examples of enediones. Therefore, a brief screen of conditions was carried out to evaluate the feasibility of such a reaction (Table 1). Firstly, our recently developed ligand- and base-free conditions for cyclohexenone derivatives18a,j,k failed to promote oxidative Heck coupling of cyclopentene-1,3-dione 1a and arylboroxine 3a (entry 1, Table 1). We thus turned to conditions using N-ligands. While oxidative Heck reactions on simple cyclohexenones using molecular oxygen19 as the oxidant have been reported to proceed at room temperature using 1,10-phenanthroline ligand 4,17 cyclopentene-1,3-dione 1a produces only trace amounts of desired oxidative Heck product 2aa at RT (entry 2) and requires higher temperatures (70 °C) for good conversion to 2aa (entry 3). A control reaction without ligand also gives poor conversion (<10%, entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Pd(II) cat. | Temp. (°C) | Result |
| a Arylboronic acid (2 equiv.) is heated under vacuum to generate the arylboroxine prior to use. b DMSO used as solvent, Pd(OTf)2 formed in situ using 5 mol% Pd(OAc)2 and 9.9 mol% TfOH. c 48 h. d Isolated yields. e 72 h. | ||||
| 1b,c | — | Pd(OTf)2 | 70 | No reaction |
| 2c |

|
Pd(OAc)2 | RT | Trace 2, conjugate addition main product |
| 3e | 4 | Pd(OAc)2 | 70 | 77% yieldd |
| 4e | — | Pd(OAc)2 | 70 | <10% conv. |
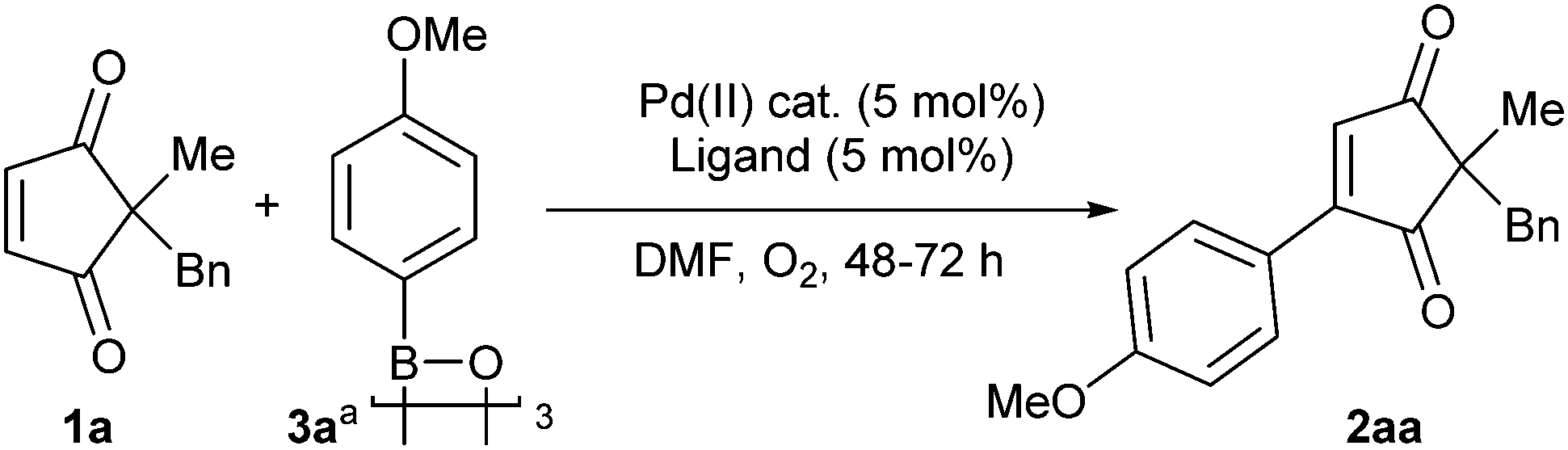
With the optimal conditions (entry 3, Table 1) in hand, a screen of cyclopentene-1,3-diones 1 was carried out (Table 2). Firstly, changing the benzyl group in 1a to a bulkier naphthyl equivalent (1b) is not detrimental to the yield (77% vs. 76% respectively, entries 1 and 2). Replacing the benzyl in 1a with an alkyl chain (1c), or with various aryls (1d–1h) are also tolerated (56–95%, entries 3–5). Next, substrates with more functionality were probed. The oxidative Heck reaction with 1i and 1j demonstrate that benzyl protected alcohols as well as esters are well tolerated (63% and 94%, entries 6 and 7). Pleasingly, even an unprotected carboxylic acid functionality is very well tolerated (83% 2ak, entry 7) as is a heterocycle (70% 2al, entry 8). These examples demonstrate that protecting groups are not always necessary for the oxidative Heck reaction. Spirocyclic 1m also reacts well (82%, entry 9). The reaction does not, however, quite tolerate enolisable protons at the 2-position of the cyclopentene-1,3-dione (1n). Instead of the desired 2an, the unexpected product 5an is observed instead, with two additional aryls installed (entry 10).20–22
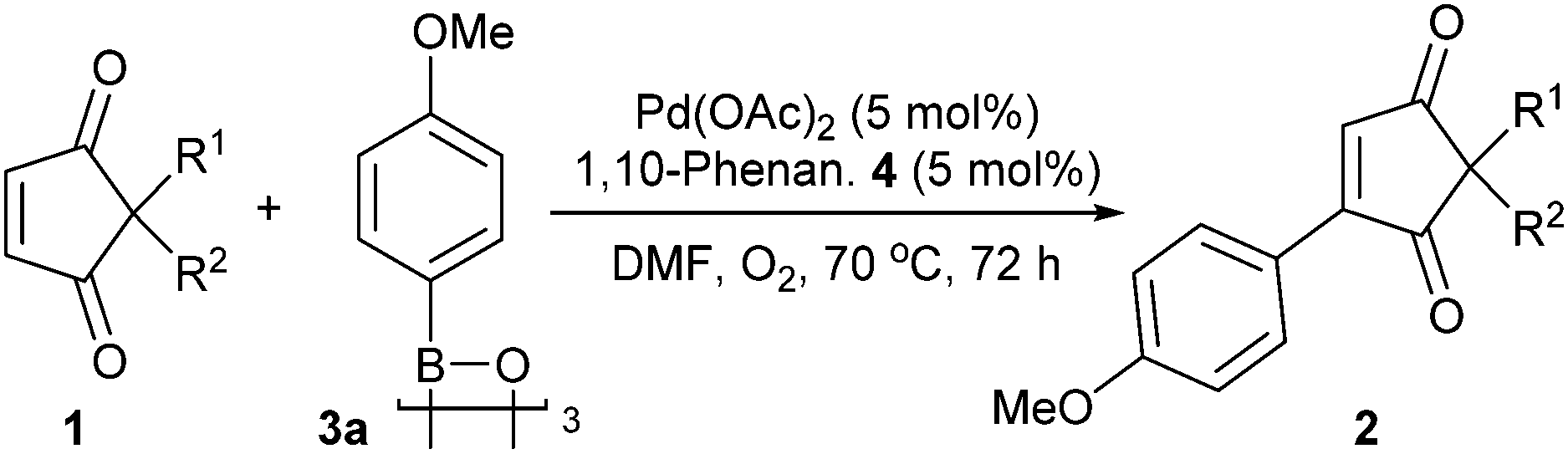



















Next, the arylboroxine scope was investigated. It should be noted that heating the commercial arylboronic acids23 under vacuum to dehydrate them to the corresponding arylboroxine prior to use provides much improved yields (e.g. 89% 2gaTable 3vs. 30% using arylboronic acid).24 The reaction conditions used so far also had to be modified in order to obtain good yields across a wider spectrum of aryl coupling partners. Portion-wise addition of the catalyst and ligand was found to be ideal for better conversions (see ESI†). Using these conditions, the arylboroxine substrate scope study shows that a wide variety of arylboroxines are suitable coupling partners (Table 3). Electron-withdrawing (2ca–2ea) as well as electron-donating substituents (2aa, 2fa–2ja) are all tolerated well under the general reactions conditions as are ortho (2fa), meta (2da, 2ga) and para substituents (2aa, 2ca, 2ea, 2ga–2ja). Once again, tolerance to unprotected functional groups such as ketone (2ea), phenol (2ha), alcohol (2ia) and amide (2ja) is demonstrated. Furthermore, the ester, chloro and unprotected hydroxyl groups in 2ca, 2da and 2ha–2ia respectively also provide a handle for further functionalisation. Polycyclic aromatic groups (2ka, 2la), including 2-fluorene with a readily oxidisable position (2la) are also pleasingly tolerated.


Finally, initial attempts at enantioselective desymmetrisation using commercially available chiral PyOX ligands 6a25 or 6b26 produced very promising results (Table 4). In order to avoid issues with competitive ligation from DMF solvent,10d DMA was used as the solvent instead27 and a lower temperature of 50 °C was also employed. To our delight, aryl substituted 1d–g and naphthyl substituted 1h substrates are desymmetrised in 74:26 to 94:6 er and excellent yields (85–100%) under these initial conditions, using both electron-donating (3a, 3h) and -withdrawing (3m) substituted aryl boroxines, thereby showing the promise and validity of our proposed idea in Scheme 1. A current limitation is that the er is modest when R is not an aryl substituent (e.g. Bn in 1a, giving 65:35 er 2aa).

In order to ascertain the absolute stereochemistry of 2 by comparison with a known structure, a one-step synthesis of preussidone4 was attempted from 1o. To our delight, (+)-preussidone was successfully obtained in 79% yield and 85:15 er, without the need for OH protection (Scheme 2).28 By comparison with literature values,4 the S stereochemistry can be assigned for 2on and thereby by analogy, also for the products in Table 4.
 | ||
| Scheme 2 Synthesis of (+)-preussidone. | ||
In conclusion, oxidative Heck couplings have been developed for 2,2-disubstituted cyclopentene-1,3-diones 1 for the first time. These substrates were found to be more challenging oxidative Heck coupling partners compared to simple alkenes or cyclohexenones, as evidenced by the higher reaction temperatures (50–70 °C vs. RT) and stricter requirements for the dehydrated arylboroxine (vs. arylboronic acid). Nevertheless, the reaction is very functional group tolerant and reacts well even in the presence of unprotected alcohols, phenols, acids, amides and ketones. Our initial enantioselective results show that direct oxidative Heck reactions on 2,2-disubstituted cyclopentene-1,3-diones is potentially a powerful method to desymmetrise all-carbon quaternary centres on the cyclopentenedione core (up to 94:6 er and quant. yields), as exemplified by the synthesis of (+)-preussidone. Further investigations into this enantioselective method are currently underway and will be reported in due course.
We thank James Jordan-Hore for preliminary experiments, EPSRC (SEW), and Erasmus (PN) for funding, the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services and Johnson Matthey for loan of Pd(OAc)2.
Notes and references
- (a) T. Hirose, T. Sunazuka, T. Shirahata, D. Yamamoto, Y. Harigaya, I. Kuwajima and S. Omura, Org. Lett., 2002, 4, 501 CrossRef CAS PubMed; (b) T. Hirose, T. Sunazuka, D. Yamamoto, E. Kaji and S. Omura, Tetrahedron Lett., 2006, 47, 6761 CrossRef CAS PubMed; (c) S. Hosokawa, K. Sekiguchi, K. Hayase, Y. Hirukawa and S. Kobayashi, Tetrahedron Lett., 2000, 41, 6435 CrossRef CAS; (d) C. C. McComas, J. B. Perales and D. L. Van Vranken, Org. Lett., 2002, 4, 2337 CrossRef CAS PubMed; (e) T. Sunazuka, T. Hirose, T. Shirahata, Y. Harigaya, M. Hayashi, K. Komiyama, S. Omura and A. B. Smith III, J. Am. Chem. Soc., 2000, 122, 2122 CrossRef CAS; (f) L. Wan and M. A. Tius, Org. Lett., 2007, 9, 647 CrossRef CAS PubMed.
- H. A. Weber, D. C. Swenson, J. B. Gloer and D. Malloch, Tetrahedron Lett., 1992, 33, 1157 CrossRef CAS.
- B. Sontag, M. Rüth, P. Spiteller, N. Arnold, W. Steglich, M. Reichert and G. Bringmann, Eur. J. Org. Chem., 2006, 1023 CrossRef CAS.
- L. Du, J. B. King, B. H. Morrow, J. K. Shen, A. N. Miller and R. H. Cichewicz, J. Nat. Prod., 2012, 75, 1819 CrossRef CAS PubMed.
- (a) R. Antkowiak, W. Z. Antkowiak, I. Banczyk and L. Mikolajczyk, Can. J. Chem., 2003, 81, 118 CrossRef CAS PubMed; (b) L. Mikolajczyk and W. Z. Antkowiak, Heterocycles, 2009, 79, 423 CrossRef CAS PubMed; (c) Z.-Y. Zhou and J.-K. Liu, Nat. Prod. Rep., 2010, 27, 1531 RSC.
- For a related report on Cu(I)-catalysed desymmetrisation via a two-step conjugate addition/oxidation method, see: (a) K. Aikawa, T. Okamoto and K. Mikami, J. Am. Chem. Soc., 2012, 134, 10329 CrossRef CAS PubMed ; for direct vinylogous nucleophilic addition of deconjugated butenolides using organocatalysts, see: ; (b) M. S. Manna and S. Mukherjee, Chem. Sci., 2014, 5, 1627 RSC.
- For review on desymmetrising cyclopentanes, see: M. S. Manna and S. Mukherjee, Org. Biomol. Chem., 2015, 13, 18 CAS.
- For recent reviews on asymmetric Heck and related reactions, see: (a) D. McCartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (b) M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282 CrossRef CAS PubMed; (c) H. Li, C. H. Ding, B. Xu and X. L. Hou, Acta Chim. Sin., 2014, 72, 765 CrossRef CAS.
- See ESI† for details on substrate synthesis.
- For selected examples of intermolecular enantioselective oxidative Heck reactions, see: (a) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (b) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) S. Sakaguchi, K. S. Yoo, J. O’Neill, J. H. Lee, T. Stewart and K. W. Jung, Angew. Chem., Int. Ed., 2008, 47, 9326 CrossRef CAS PubMed; (d) K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O’Neill and K. W. Jung, Org. Lett., 2007, 9, 3933 CrossRef CAS PubMed.
- For reviews of catalytic enantioselective desymmetrisations, see: (a) K. Mikami and A. Yoshida, J. Synth. Org. Chem., Jpn., 2002, 60, 732 CrossRef CAS; (b) T. Rovis, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, Hoboken, NJ, 2007, p. 275 Search PubMed; (c) R. S. Ward, Chem. Soc. Rev., 1990, 19, 1 RSC; (d) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765 RSC.
- Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Blackwell Science Publ, Oxford, 2005 Search PubMed.
- M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130 CrossRef CAS PubMed.
- (a) D. Tanaka and A. G. Myers, Org. Lett., 2004, 6, 433 CrossRef CAS PubMed; (b) Y. Fall, H. Doucet and M. Santelli, Tetrahedron, 2009, 65, 489 CrossRef CAS PubMed.
- For reviews on oxidative Heck, see: (a) B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 CrossRef CAS PubMed; (b) Y. J. Su and N. Jiao, Curr. Org. Chem., 2011, 15, 3362 CrossRef CAS.
- Selected papers on oxidative Heck, for examples on cyclic enones see ref. 17 and 18: (a) M. M. S. Andappan, P. Nilsson and M. Larhed, Chem. Commun., 2004, 218 RSC; (b) C. S. Cho and S. Uemura, J. Organomet. Chem., 1994, 465, 85 CrossRef CAS; (c) J. D. Crowley, K. D. Hanni, A.-L. Lee and D. A. Leigh, J. Am. Chem. Soc., 2007, 129, 12092 CrossRef CAS PubMed; (d) J. H. Delcamp, A. P. Brucks and M. C. White, J. Am. Chem. Soc., 2008, 130, 11270 CrossRef CAS PubMed; (e) J. H. Delcamp, P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 8460 CrossRef CAS PubMed; (f) X. Du, M. Suguro, K. Hirabayashi, A. Mori, T. Nishikata, N. Hagiwara, K. Kawata, T. Okeda, H. F. Wang, K. Fugami and M. Kosugi, Org. Lett., 2001, 3, 3313 CrossRef CAS PubMed; (g) Z. He, S. Kirchberg, R. Froehlich and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 3699 CrossRef CAS PubMed; (h) A. Inoue, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 2003, 125, 1484 CrossRef CAS PubMed; (i) Y. C. Jung, R. K. Mishra, C. H. Yoon and K. W. Jung, Org. Lett., 2003, 5, 2231 CrossRef CAS PubMed; (j) L. Meng, C. Liu, W. Zhang, C. Zhou and A. Lei, Chem. Commun., 2014, 50, 1110 RSC; (k) J. Ruan, X. Li, O. Saidi and J. Xiao, J. Am. Chem. Soc., 2008, 130, 2424 CrossRef CAS PubMed; (l) Y. Su and N. Jiao, Org. Lett., 2009, 11, 2980 CrossRef CAS PubMed; (m) P. Sun, Y. Zhu, H. Yang, H. Yan, L. Lu, X. Zhang and J. Mao, Org. Biomol. Chem., 2012, 10, 4512 RSC; (n) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 13981 CrossRef CAS PubMed; (o) C. Zheng, D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2012, 134, 16496 CrossRef CAS PubMed.
- K. S. Yoo, C. H. Yoon and K. W. Jung, J. Am. Chem. Soc., 2006, 128, 16384 CrossRef CAS PubMed.
- For example, see: (a) S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A.-L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876 CrossRef CAS PubMed; (b) Y. W. Kim and G. I. Georg, Org. Lett., 2014, 16, 1574 CrossRef CAS PubMed; (c) A. Carrër, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef PubMed; (d) A. L. Gottumukkala, J. F. Teichert, D. Heijnen, N. Eisink, S. van Dijk, C. Ferrer, A. van den Hoogenband and A. J. Minnaard, J. Org. Chem., 2011, 76, 3498 CrossRef CAS PubMed; (e) Y. Izawa, C. Zheng and S. S. Stahl, Angew. Chem., Int. Ed., 2013, 52, 3672 CrossRef CAS PubMed; (f) Y. Li, Z. Qi, H. Wang, X. Fu and C. Duan, J. Org. Chem., 2012, 77, 2053 CrossRef CAS PubMed; (g) B. Mondal, S. Hazra and B. Roy, Tetrahedron Lett., 2014, 55, 1077 CrossRef CAS PubMed; (h) D.-C. Xiong, L.-H. Zhang and X.-S. Ye, Org. Lett., 2009, 11, 1709 CrossRef CAS PubMed; (i) K. S. Yoo, J. O’Neill, S. Sakaguchi, R. Giles, J. H. Lee and K. W. Jung, J. Org. Chem., 2010, 75, 95 CrossRef CAS PubMed; (j) J. A. Jordan-Hore, J. N. Sanderson and A.-L. Lee, Org. Lett., 2012, 14, 2508 CrossRef CAS PubMed; (k) S. E. Walker, J. Boehnke, P. E. Glen, S. Levey, L. Patrick, J. A. Jordan-Hore and A.-L. Lee, Org. Lett., 2013, 15, 1886 CrossRef CAS PubMed . See also ref. 16a and 17.
- K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854 RSC.
- One possible explanation for this is that tautomerisation of the desired oxidative Heck product can now occur, allowing 1,2-addition (see ref. 21)/tautomerisation/coupling via π-allyl intermediate (see ref. 22) to produce the triarylated product 5an.
- T. Yamamoto, T. Ohta and Y. Ito, Org. Lett., 2005, 7, 4153 CrossRef CAS PubMed.
- Y. Kayaki, T. Koda and T. Ikariya, Eur. J. Org. Chem., 2004, 4989 CrossRef CAS.
- It should be noted that commercial aryl boronic acid samples are often a mixture of the aryl boronic acid and arylboroxine in equilibrium. D. G. Hall, Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–133 Search PubMed.
- Alternatively, arylboronic acid pinacol esters can also be used, although the yields are slightly lower than with arylboronic acids. For example, 2bh is formed in 84% and 78% yields using phenylboronic acid and pinacol ester respectively, under general conditions shown in Table 2.
- H. Shimizu, J. C. Holder and B. M. Stoltz, Beilstein J. Org. Chem., 2013, 9, 1637 CrossRef PubMed.
- E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed.
- R. Díaz-Torres and S. Alvarez, Dalton Trans., 2011, 40, 10742 RSC.
- The arylboronic acid pinacol ester 7n was used here as it is commercially available while the corresponding arylboronic acid is not.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR and 13C NMR spectra and full characterisation of new compounds. See DOI: 10.1039/c5cc00407a |
| This journal is © The Royal Society of Chemistry 2015 |