
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of nanodiamonds from carbon nanoparticles at atmospheric pressure†
Ali Reza
Kamali
* and
Derek J.
Fray
Materials Science and Metallurgy Department, University of Cambridge, 27 Charles Babbage Road, Cambridge, CB3 0FSA, UK. E-mail: ark42@cam.ac.uk; djf25@cam.ac.uk
First published on 23rd January 2015
Abstract
A route for producing diamond nanocrystals is reported in this paper. Li2CO3 containing carbon nanostructures synthesised in molten LiCl were transformed to nanodiamonds by simple heating at atmospheric pressure, far less severe conditions than conventional processes. The method presented offers the possibility of bulk production.
Elemental carbon can exist in diverse forms such as graphite, diamond and fullerene owing to its ability to form sp, sp2 and sp3 hybridized bonds and the transformation of graphite to diamond has been subject to a large number of studies.1–14 The phase diagram of carbon shows that diamond is the stable phase if the pressure is beyond 1.7 GPa at room temperature1 although, once formed, it is stable under ambient conditions. Generally, the conversion of graphite to diamond requires extremely high temperatures (>1000 °C) and pressures (>15 GPa). In the 1950s, it was found that graphite dissolves in molten transition metals, such as iron, cobalt and nickel and then precipitates as diamond at around 5–6 GPa and 1300–1700 °C.2 Non-metallic catalysts such as inorganic salts have been used although higher P–T conditions are required as well as longer reaction times.3–7 Using alkali carbonates it was found that the yield was related to the cation radius in the sequence Li2CO3 > Na2CO3 > K2CO3 > Cs2CO3.4 Furthermore, it is known that carbonates are found as inclusions in natural diamonds8,9 and, also, diamonds occur in carbonate-bearing and carbonate rich rocks.10 At the pressure of 7 GPa and the temperature of 1700 °C, Pal'yanov found that the required time for diamonds to nucleate and grow was 2 hours.4 Nanostructured diamond particles have been known since the 1960s and have been produced by shock wave compression of graphite and carbon black mixed with a catalyst.12 An alternative method is to use a mixture of carbon and high energy explosives or to utilise the carbon contained in high energy explosives. These diamonds are known as detonation nanodiamonds (DND).13 Other methods to produce nanodiamonds have used microwave plasma torches14 and HPHT conditions.15 All the methods outlined above require extreme conditions and are poorly suited for mass production, which is unfortunate as nanodiamonds have remarkable properties which could find myriad applications in biomedicine, nanocomposites and as seeding material for the growth of diamond films. A two-step process for the synthesis of nanodiamonds is described in this paper.
Theoretical analyses have shown that sp3 diamond nucleation from sp2 carbon is preferable inside a carbon nanotube (CNT) or nanoparticle due to the effect of the surface tension brought about by the nanometre-sized curvature of carbon nanomaterials.11 Furthermore, sp3 bonds have been observed in used graphitic anodes retrieved from lithium batteries in which lithium has been inserted into and extracted from the graphite over many cycles.16 There have, therefore, been many attempts to convert CNTs into diamond using laser irradiation, shock waves, spark plasma sintering and radio-frequency hydrogen plasma techniques.17–25 Some success was achieved at 4.5 GPa and 1300 °C with a Ni–Mn–Co catalyst where it was observed that the CNTs first transformed into quasi-spherical onion-like particles. Diamond crystals could then be nucleated from the onion-like particles with the assistance of the catalyst. Similar results were obtained with a Fe–Ni catalyst. All the CNTs that have been investigated so far have been produced by chemical vapour deposition (CVD) with the aid of catalysts.
Another route of CNT and nanoparticle synthesis uses the intercalation of lithium from molten lithium chloride into graphite by electrolysis.26 The mechanism by which this occurs is that the lithium ions discharge on the cathode and pass into the graphite between the layers of graphene under the influence of the cathodic potential. The electrolysis reaction can be expressed as:
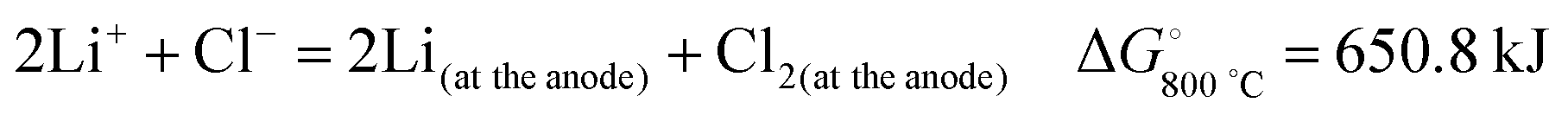 | (1) |
Oxygen anions formed in the molten LiCl can be oxidised on the graphite anode to produce CO2, which subsequently reacts with the Li2O dissolved in the molten salt to form Li2CO3:
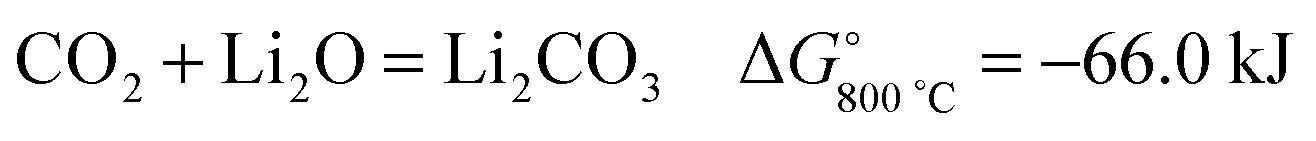 | (2) |
The micro- and nanostructure of the as-synthesised ECM is shown in Fig. 1a and b. It contains a mixture of CNTs with diameters in a wide range of 10–500 nm, spherical nanoparticles, typically less than 100 nm, and a fraction of micrometer-sized carbon particles. The CNTs and nanoparticles were examined by TEM and it appeared that inorganic materials such as lithium carbonate were trapped within the graphitic nanostructure of CNTs and nanoparticles (Fig. 2).
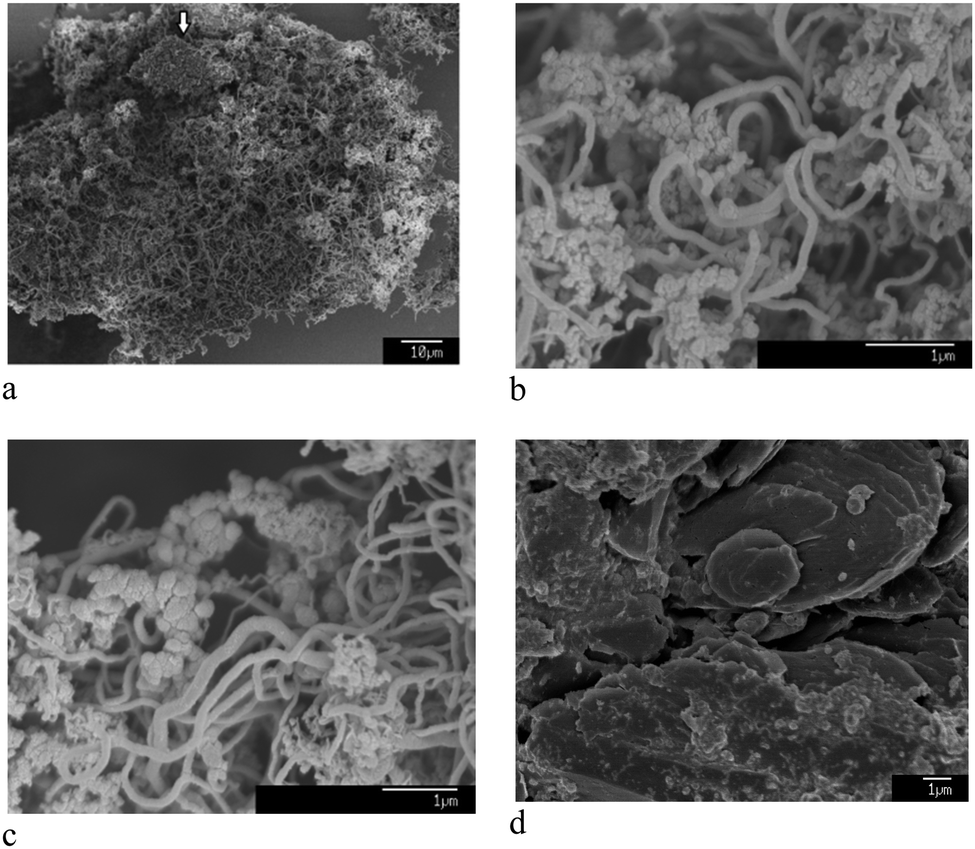 | ||
| Fig. 1 (a) and (b) show SEM morphology of the as-synthesised ECM. It contains a mixture of CNTs, carbon nanoparticles, and some micrometer-sized carbon particles such as one pointed by the arrow in (a). (c) and (d) show SEM morphology of the material heated to 400 and 570 °C, respectively, and subsequently cooled down to the room temperature under ambient air flow of 100 mL min−1 (see Fig. 3b-upper panel). | ||
 | ||
| Fig. 2 TEM micrograph of the as-synthesized ECM. The left panel shows a number of CNTs, and the inset is a typical selected area diffraction pattern confirming the presence of graphitic carbon and Li2CO3 single-crystals. The right panel exhibits two high resolution TEM images. The upper image shows a wall of a CNT demonstrating that Li2CO3 nanocrystals, pointed by arrows, embedded into the graphitic structure of the wall. The down image presents a carbon nanoparticle in which Li2CO3 nanocrystals are encapsulated in carbon shells. | ||
DSC and TG analyses of the ECM (Fig. 3b-upper panel), conducted at the rate of 20 °C min−1 under ambient air flow of 100 mL min−1, indicates the occurrence of two exothermic events with peaks maximum temperature at 511 and 637 °C. The ECM was heated to the temperatures just before and after the first exothermic peak, and then cooled down to the room temperature. Heating of ECM up to the temperatures below the first peak (for example 400 °C) didn't affect the morphology of the material, and CNTs and nanoparticles could still be clearly seen in the SEM images of the heated samples (see Fig. 1c). The sample heated to 570 °C, which is beyond the first exothermic peak, experienced a massive weight loss. Moreover, no carbon nanomaterial could be indentified in the SEM images of the heated samples (see Fig. 1d). It indicates that the first and the second exothermic peaks in DSC curve of the ECM shown in Fig. 3b-upper panel, relates to the oxidation of the nanometre sized and the micrometre sized fractions of ECM, respectively.
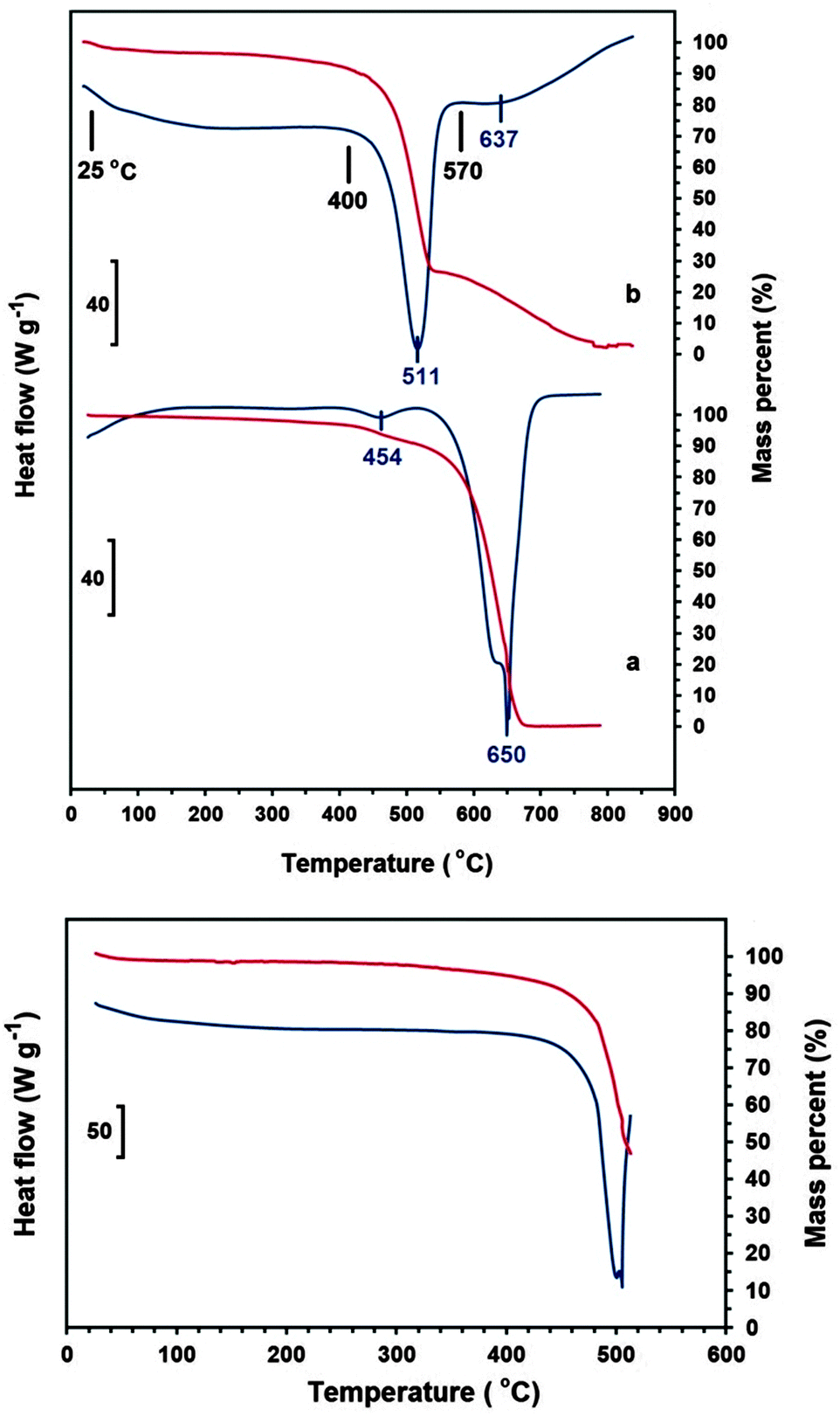 | ||
| Fig. 3 DSC (blue lines) and TG (red lines) of (upper panel-a) CVD MWCNTs and (upper panel-b) the ECM in the range of 25–800 °C. Morphology of the as-synthesised ECM and also ECM heated to 400 and 570 °C is shown in Fig. 1. The down panel shows DSC–TG curves of the ECM in the range of 25–515 °C, and the morphology of the sample obtained is exhibited in Fig. 4. The analyses were carried out under ambient air flow of 100 mL min−1. | ||
Fig. 3a-upper panel shows the DSC and TG analyses of the CVD MWCNTs heated at the rate of 20 °C min−1 under ambient air flow of 100 mL min−1. The DSC curve exhibits one small exothermic peak at 454 °C, which is related to the oxidation of the amorphous fraction of the sample, followed by a large exothermic peak at 650 °C corresponding to the oxidation of the whole remaining material. It is known that the oxidation temperature of carbon nanomaterials depends on their degree of graphitisation; the greater crystallinity, the higher oxidation temperature.32 However, despite its higher degree of crystallinity, the ECM exhibits a lower oxidation temperature than the CVD MWCNTs. It can be explained by the presence of Li2CO3 nanocrystals in the nanostructure of ECM, which can catalyse the oxidation of the material at lower temperatures, as explained in more details elsewhere.33
It is known that molten carbonates of alkali and alkaline metals are able to act as solvent-catalyst for diamond formation from graphite at typical HPHT conditions of 5 to 8 GPa and 1600 to 2150 °C.4 Subsequently, several other inorganic melts, including metal halides such as LiCl and multicomponent systems, have also been shown to catalyse the conversion of graphite to diamond at very high temperatures and pressures.5 The presence of nanometre-sized catalyst crystals embedded in the graphitic nanostructure of the electrolytic carbon materials makes it attractive for the production of diamond.
According to Fig. 3b-upper panel, the oxidation of CNTs and nanopaticles happens at the temperature window of 420–550 °C. It was observed that if the ECM is heated to specific temperatures within this oxidation window, the carbon nanomaterials are transformed into nanodiamonds. For example, as shown in Fig. 3-down panel, the ECM was heated to 515 °C, and then cooled down to the room temperature. The (111) reflection of diamond can be seen in the XRD spectrum of the partially oxidised material shown in Fig. 1S(c) (ESI†).
The morphology of the sample heated to 515 °C was examined by SEM, which showed that the sample has been mainly converted to octagonal nanodiamonds from 5 nm to 1 μm (Fig. 4, the left and middle panels). The right panel in Fig. 4 shows diamond crystals growing on a large carbon particle of about 20 μm in size. The position of a diamond crystal is marked by an arrow. The large carbon particle is likely to be formed by sintering of carbon nanostructures during combustion of the electrolytic carbon material.
 | ||
| Fig. 4 SEM micrographs of the ECM heated to 515 °C in air, showing nano and micron sized diamonds. The right hand panel shows a number of diamond crystals which are growing on a carbon substrate, one of which is pointed by the arrow. | ||
Further confirmation of the diamond structure was obtained by examining the sample by TEM where it was seen that the electron diffraction pattern corresponded to the (111) plane of diamond (Fig. 5). The presence of diamond was also confirmed by the Raman spectrum (Fig. 6) (for more details see the ESI†). It is worthy to mention that the quality of the nanodiamonds produced is much higher than those obtained by detonation synthesis. The detonation nanodiamonds are so vague in shape that their SEM image never gave discernible shapes, such as shown in Fig. 4.
 | ||
| Fig. 5 TEM micrograph of the ECM after heating in air to 515 °C. The electron diffraction pattern taken from a selected area of the TEM micrograph (inset) originates from the (111) plane of diamond in the cubic structure. | ||
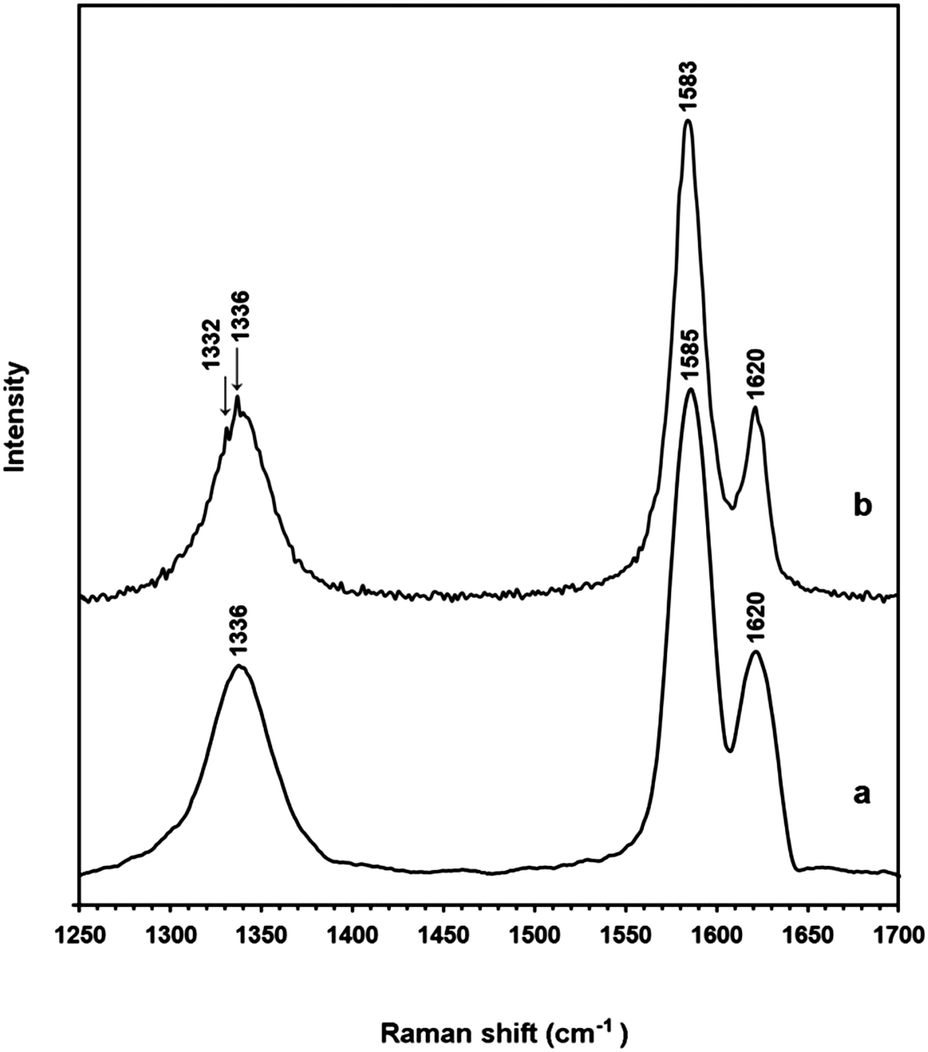 | ||
| Fig. 6 Raman spectra of (a) the as-synthesised ECM and (b) the ECM heated in air to 515 °C (see Fig. 3-down panel) showing the peak of diamond at 1332 cm−1. | ||
After heat treatment it has been observed that the true density of ECM increases from 2.2 g cm−3 to 3.0 g cm−3, indicating the phase transition from graphite to diamond as the molar volume of diamond is 3.42 cm3 compared to that of graphite which is 5.34 cm3.
We assert that by producing carbon nanomaterials electrochemically by intercalation, lithium salts are incorporated between the graphene sheets and these are able to catalyse the transformation to diamond by simply heating in air at atmospheric pressure. It should be noted that although the CNTs and nanoparticles are ignited at about 420 °C, registered in the DSC curve of Fig. 3-down panel, the true local temperature of the carbon nanomaterials during the oxidation is likely to reach as high as 4500 °C (for more details see the ESI†). At this temperature, the Li2CO3 encapsulated in graphitic shells is also likely to produce a considerable amount of internal pressure. Further study is required for better understanding the mechanism involved.
This process should be much cheaper than that produced by HPHT and should be scalable to produce a more economical product. The applied voltages and current densities and rate of production of carbon nanotubes in molten LiCl34 are very similar to that of aluminium in the Hall–Heroult cells which produce 45 M tonnes per annum worldwide. Upscaling should not be problem. The cost of aluminium is around $2 kg−1 whilst the present price of nanodiamonds is about $3 g−1! Using this novel technique should result in a substantial drop in the cost of nanodiamonds and a widening of the applications.
Notes and references
- F. P. Bundy, J. Geophys. Res., 1980, 85(B12), 6930 CrossRef CAS.
- H. P. Bovenkerk, F. P. Bundy, H. T. Hall, H. M. Strong and J. R. H. Wentorf, Nature, 1959, 184, 1094 CrossRef CAS.
- M. Akaishi, H. Kanda and S. Yamaoka, J. Cryst. Growth, 1990, 104, 578 CrossRef CAS.
- Y. N. Pal'yanov, A. G. Sokol, Y. M. Borzdov, A. F. Khokhryakov, A. F. Shatsky and N. V. Sobolev, Diamond Relat. Mater., 1999, 8, 1118 CrossRef.
- Y. Wang and H. Kanda, Diamond Relat. Mater., 1998, 7, 57 CrossRef CAS.
- E. Tomlinson, A. Jones and J. Milledge, Lithos, 2004, 77, 287 CrossRef CAS PubMed.
- A. V. Spivak, Y. A. Litvin, A. V. Shushkanova, V. Y. Litvin and A. A. Shiryaev, Eur. J. Mineral., 2008, 20, 341 CrossRef CAS.
- A. V. Krasheninnikov and F. Banhart, Nat. Mater., 2007, 6, 723 CrossRef CAS PubMed.
- L. T. Sun, J. L. Gong, Z. Y. Zhu, D. Z. Zhu, S. X. He and Z. X. Wang, Appl. Phys. Lett., 2004, 84, 2901 CrossRef CAS PubMed.
- F. Banhart and P. M. Ajayan, Nature, 1996, 382, 433 CrossRef CAS.
- Q. X. Liu, C. X. Wang, S. W. Li, J. X. Zhang and G. W. Yang, Carbon, 2004, 42, 629 CrossRef CAS PubMed.
- P. S. Decarli and J. C. Jamieson, Science, 1961, 133, 1821 CAS.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11 CrossRef CAS PubMed.
- F. Fendrych, A. Taylor, L. Peksa, I. Kratochvilova, J. Vlcek, V. Rezacova, V. Petrak, Z. Kluiber, L. Fekete, M. Liehr and M. Nesladek, J. Phys. D: Appl. Phys., 2010, 43, 374018 CrossRef.
- M. Mandal, F. Haso, T. Liu, Y. Fei and K. Landskron, Chem. Commun., 2014, 50, 11307 RSC.
- Q. Jiao, X. Zhu, X. Xiao, X. Zuo, J. Nan and L. Wang, ECS Electrochem. Lett., 2013, 2, 1127 Search PubMed.
- B. Wei, J. Zhang, J. Liang, W. Liu, Z. Gao and D. Wu, J. Mater. Sci. Lett., 1997, 16, 402 CrossRef CAS.
- B. Wei, J. Zhang, J. Liang and D. Wu, Carbon, 1998, 16, 997 CrossRef.
- W. Han, S. Fan, Q. Li and C. L. Zhang, Jpn. J. Appl. Phys., Part 2, 1998, 37, L1085 CAS.
- L. Cao, C. Gao, H. Sun, G. Zou, Z. Zhang, X. Zhang, M. He and M. Zhang, Carbon, 2001, 39, 311 CrossRef CAS.
- H. Yusa, Diamond Relat. Mater., 2002, 11, 87 CrossRef CAS.
- Y. Q. Zhu, T. Sekine, T. Kobayashi, E. Takazawa, M. Terrones and H. Terrones, Chem. Phys. Lett., 1998, 287, 689 CrossRef CAS.
- A. Merlen, P. Toulemonde, S. L. Floch, G. Montagnac, T. Hammouda, O. Marty and A. San Miguel, Carbon, 2009, 47, 1643 CrossRef CAS PubMed.
- F. Zhang, M. Adam, F. Ahmed, E. Otterstein and E. Burkel, Diamond Relat. Mater., 2011, 20, 853 CrossRef CAS PubMed.
- F. Zhang, F. Ahmed, G. Holzhuter and E. Burkel, J. Cryst. Growth, 2012, 340, 1 CrossRef CAS PubMed.
- W. K. Hsu, J. P. Hare, M. Terrones, H. W. Kroto, D. R. M. Walton and P. J. F. Harris, Nature, 1995, 377, 687 CrossRef CAS.
- C. Schwandt, A. T. Dimitrov and D. J. Fray, J. Electroanal. Chem., 2010, 647, 150 CrossRef CAS PubMed.
- A. R. Kamali, C. Schwandt and D. J. Fray, Mater. Charact., 2011, 62, 987 CrossRef CAS PubMed.
- Y. Sakamura, J. Electrochem. Soc., 2010, 157, E135 CrossRef CAS PubMed.
- A. R. Kamali, D. J. Fray and C. Schwandt, J. Therm. Anal. Calorim., 2011, 104, 619 CrossRef CAS PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102 CrossRef CAS.
- R. Brukh and S. Mitra, J. Mater. Chem., 2007, 17, 619 RSC.
- A. R. Kamali, C. Schwandt and D. J. Fray, Corros. Sci., 2012, 54, 307 CrossRef CAS PubMed.
- A. R. Kamali and D. J. Fray, Carbon, 2014, 77, 835 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc00233h |
| This journal is © The Royal Society of Chemistry 2015 |